Clinical investigations in Northern Ireland
How manufacturers in Northern Ireland should undertake a clinical investigation for medical devices to ensure they comply with EU medical device regulations.
Applies to Northern Ireland
This guidance is for manufacturers undertaking a clinical investigation in Northern Ireland.
The rules for notifying the MHRA of a clinical investigation in Northern Ireland differ from those applicable to Great Britain (England, Wales and Scotland). You should also read our guidance on clinical investigations for medical devices in Great Britain as much of it still applies.
This guidance explains the additional requirements under the EU legislation.
The Northern Ireland Protocol requires Northern Ireland to continue to align with EU rules for devices as set out in the Medical Device Regulation (EU) 2017/745 (EU MDR) and the In Vitro Diagnostic Medical Device Regulation (EU) 2017/746 (EU IVDR).
Clinical investigations being conducted in Northern Ireland must be submitted for approval in accordance with these regulations.
Where a clinical investigation includes sites in both Great Britain and Northern Ireland, submission to the MHRA must be made in line with the requirements of the EU MDR. By meeting the EU MDR, requirements of the UK MDR for clinical investigations are deemed to be satisfied. Therefore, a single application made to the MHRA under the EU MDR will cover any sites proposed in both Great Britain and Northern Ireland for the same clinical investigation.
When a clinical investigation is required
You must notify the MHRA of your planned clinical investigation, if you plan on using a non-UKCA or non-CE marked medical device on human participants and an exemption does not apply.
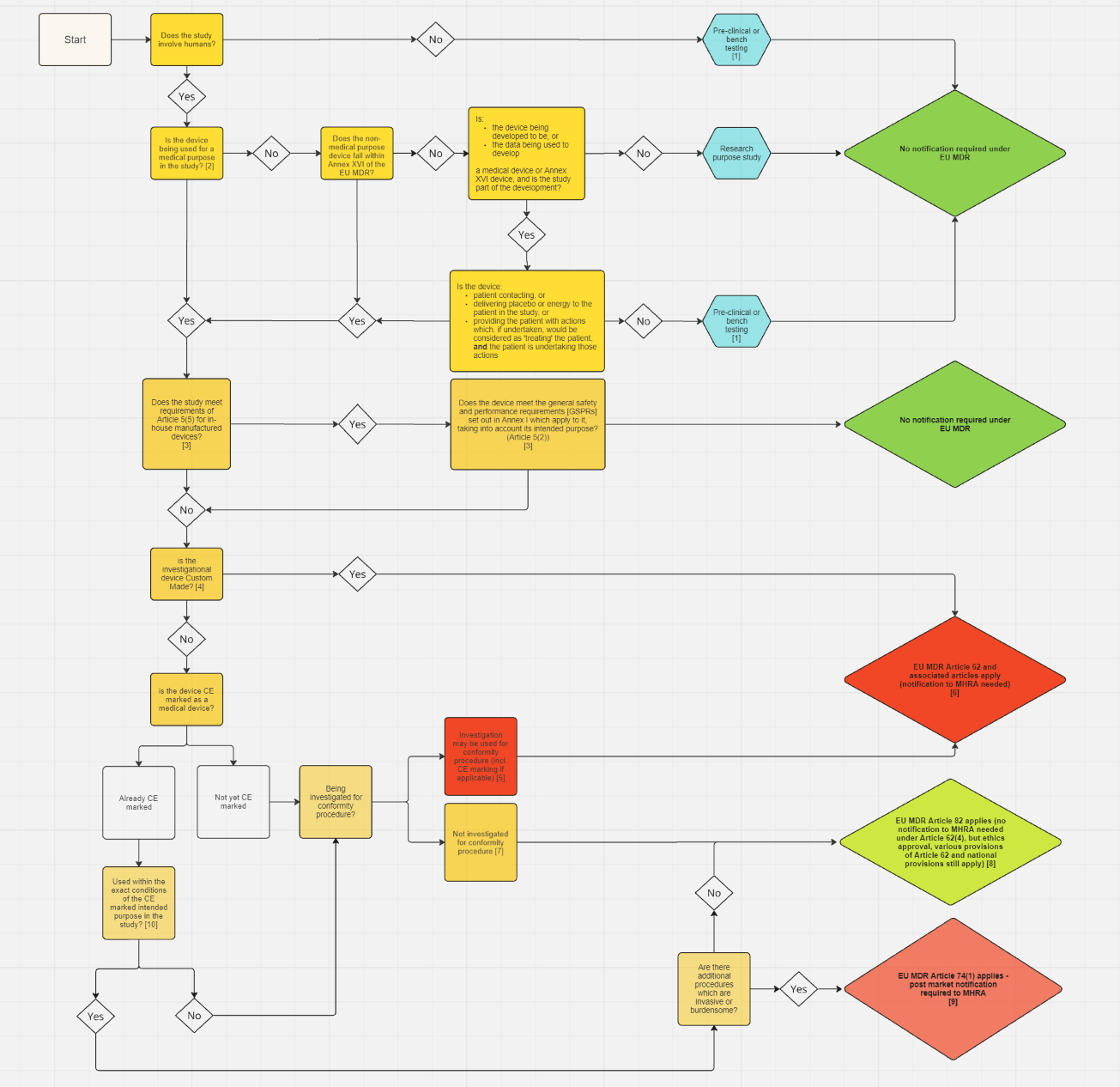
For studies in Northern Ireland, please use this flow chart and accompanying guidance to determine if you need to submit an application to MHRA.
{kind=link}
A clinical investigation of a non-UKCA or CE marked medical device should at least be considered in the following circumstances:
- the device is an implantable or Class III medical device
- the introduction of a completely new concept of device into clinical practice where components, features and/or methods of action, are previously unknown
- where an existing device is modified in such a way that it contains a novel feature particularly if such a feature has an important physiological effect; or where the modification might significantly affect the clinical performance or safety of the device
- where a device incorporates materials previously untested in humans, coming into contact with the human body or where existing materials are applied to a new location in the human body or where the materials are to be used for a significantly longer time than previously, in which case compatibility and biological safety will need to be considered
- where a device, either UKCA or CE marked or non-UKCA or CE marked, is proposed for a new purpose or function
- where in vitro or animal testing of the device cannot mimic the clinical situation
- where there is a new manufacturer especially of a high-risk device.
However, the EU MDR explicitly states that in the case of implantable devices and class III devices, clinical investigations shall be performed, except if:
- the device has been designed by modifications of a device already marketed by the same manufacturer or
- the modified device has been demonstrated by the manufacturer to be equivalent to the marketed device and
- the clinical evaluation of the marketed device is sufficient to demonstrate conformity of the modified device with the general safety and performance requirements (GSPRs)
Where the manufacturer of the modified device is not the manufacturer of the marketed device, then in addition to the above bullet points the following must also be in place:
- the two manufacturers must have a contract in place that allows the manufacturer of the modified device full access to the technical documentation of the marketed device on an ongoing basis
- the original clinical evaluation for the marketed device has been performed in compliance with Regulation 2017/745.
Clinical investigations are also not required for implantable and class III devices:
- which have been lawfully placed on the market or put into service before 26th May 2021 in accordance with Directive 90/385/EEC or Directive 93/42/EEC
- the clinical evaluation is based on sufficient clinical data and complies with relevant product-specific common specification where available.
or
- are sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips or connectors
- the clinical evaluation is based on sufficient clinical data and complies with relevant product-specific common specification where available
Notifying the MHRA of your planned investigation
All notifications should be submitted using the application process on the Integrated Research Application System (IRAS) portal.
The clinical investigation application form and submission checklist on IRAS help the MHRA record the applications and accompanying documentation, and applicants ensure that all required information is available and referenced appropriately.
You can find out more about how to compile your application and the process in our guidance on making a submission. This includes information on what you need to include.
Before devices intended for clinical investigation in Northern Ireland are made available to a medical practitioner for the purposes of clinical investigation, the sponsor of the clinical investigation (or their legal representatives in the European Union) must give up to 65 days of prior notice to the Secretary of State for Health by writing to the UK competent authority (the MHRA).
The MHRA will review the clinical investigation during the first 45 days and will issue you with a final decision of either authorisation or refusal. The MHRA may extend the 45-day review period by a further 20 days should an expert assessor be required to review the application. We will notify you of any extension by email.
The clinical investigation may not be conducted without authorisation by the MHRA. We may refuse authorisation if the clinical investigation does not meet the requirements of the regulation, the application is incomplete, the device or the clinical investigation plan and investigators brochure do not correspond to state of scientific knowledge and do not provide evidence for safety, performance or benefit of the device (Regulation 2017/745 Article 71.4).
You can find out more about how to notify us of a planned clinical investigation.
For studies with sites in Northern Ireland, a representative may need to be appointed in Northern Ireland or the EU.
You can find out more in our guidance on regulating medical devices in Northern Ireland.
Complying with clinical investigation requirements
Unless an exemption applies, all medical devices to be used on humans must be UKCA or CE marked for the purpose that they are being used for, unless they are being used as part of a clinical investigation designed to investigate the performance and safety of the medical device or accessory.
In order to be able to UKCA or CE mark any device, a manufacturer must demonstrate that the stated device complies with the relevant essential requirements as listed in Part II and Part III of the UK MDR, Annex I (as modified by Schedule 2A to the UK MDR) or the general safety and performance requirements (GSPRs) in Annex I of the EU MDR.
To demonstrate such compliance, it will usually be necessary to provide clinical data, which can consist of a critical evaluation of:
- the relevant scientific literature currently available relating to the safety, performance, design characteristics and intended purpose of the device, where there is demonstration of equivalence of the device to the device to which the data relates and the data adequately demonstrates compliance with the relevant essential requirements and general safety and performance requirements (GSPRs)
- the results of all the clinical investigations made
- the combined data of the relevant scientific literature and results of the clinical investigation
Critical analysis and evaluation of scientific literature are broad concepts which can take account of the experience of the device in question or of an established device which is already on the market and used in clinical practice and with which equivalence can be demonstrated in terms of technology, critical performance, design, principles of operation, biological safety, population involved, conditions of use and clinical purpose.
When UKCA or CE marking a device, unless safety and performance can be adequately demonstrated by other means, data generated from a specifically designed clinical investigation of a medical device are likely to be required, in particular with implantable and class III devices.
Clinical investigations must be designed to establish and verify:
- that, under normal conditions of use, a device is designed, manufactured and packaged in such a way that it is suitable for one or more of the specific purposes listed in point (1) of Article 2 of the EU MDR, and achieves the performance intended as specified by its manufacturer
- the clinical benefits of a device as specified by its manufacturer
- the clinical safety of the device and to determine any undesirable side effects, under normal conditions of use of the device, and assess whether they constitute acceptable risks when weighed against the benefits to be achieved by the device
The legal requirements as to methodology and ethical considerations relating to clinical investigations are set out in EU MDR (Chapter VI – Clinical Evaluation and Clinical Investigations, specifically Articles 62 – 82).
In particular, the clinical investigation must:
- be designed and conducted in such a way that the rights, safety, dignity and well-being of the subjects participating in a clinical investigation are protected and prevail over all other interests
- be designed and conducted to ensure that the clinical data generated is scientifically valid, reliable and robust
- ensure vulnerable populations and subjects are appropriately protected in accordance with Articles 64 to 68 of the regulation
- ensure the anticipated benefits to the subjects or to public health justify the foreseeable risks and inconveniences and compliance with this condition is constantly monitored
- ensure the subject or, where the subject is not able to give informed consent, his or her legally designated representative has given informed consent in accordance with Article 63
- ensure the subject or, where the subject is not able to give informed consent, his or her legally designated representative, has been provided with the contact details of an entity where further information can be received in case of need
- ensure the rights of the subject to physical and mental integrity, to privacy and to the protection of the data concerning him or her in accordance with Directive 95/46/EC are safeguarded
- be designed to involve as little pain, discomfort, fear and any other foreseeable risk as possible for the subjects, and both the risk threshold and the degree of distress are specifically defined in the clinical investigation plan and constantly monitored
- ensure the medical care provided to the subjects is the responsibility of an appropriately qualified medical doctor or, where appropriate, a qualified dental practitioner or any other person qualified to provide the relevant patient care under clinical investigation conditions
- ensure no undue influence, including that of a financial nature, is exerted on the subject, or, where applicable, on his or her legally designated representatives, to participate in the clinical investigation
- involve only investigational devices that conform to the applicable general safety and performance requirements apart from the aspects covered by the clinical investigation and that, with regard to those aspects, every precaution has been taken to protect the health and safety of the subjects including where appropriate, technical and biological safety testing and preclinical evaluation, as well as provisions in the field of occupational safety and accident prevention, taking into consideration the state of the art
- ensure that any subject or, where the subject is not able to give informed consent, his or her legally designated representative may, without any resulting detriment and without having to provide any justification, withdraw from the clinical investigation at any time by revoking his or her informed consent
- ensure the investigator qualifies for the role of investigator by having the necessary scientific knowledge and experience in patient care
- ensure that other personnel involved in conducting a clinical investigation shall be suitably qualified, by education, training or experience in the relevant medical field and in clinical research methodology, to perform their tasks
- be conducted at facilities that are suitable for the clinical investigation and shall be similar to the facilities where the device is intended to be used
Special circumstances requiring particular data
Change in the intended use or performance claims of a device
Clinical data may be required in the case of a device already authorised to carry the UKCA or CE marking where that device is to be used for a new purpose and eventually UKCA or CE marked for that new purpose. The clinical data may need to be generated by a specifically designed clinical investigation, in which case a notification should be made to the MHRA.
Comparative studies
You are not usually required to notify the MHRA if a device is UKCA or CE marked for the purpose intended or, in the case of a comparative study of two devices, where each has obtained prior UKCA or CE marking, and each is used for their original purpose.
However, relevant ethics committee approval would still be required in both cases.
Where at least one of the devices under study is not UKCA or CE marked, the manufacturers of the non-UKCA or non-CE marked devices must notify the clinical investigation to the MHRA.
Prototype devices
We recognise that a manufacturer may wish to submit a small number of ‘prototype models’ of a device to clinical investigation to assess safety or performance and that such prototypes may need to undergo changes prior to large-scale production.
These changes will be regarded as variations included within one application unless, in the view of the MHRA, the risk to patients or users is increased by the proposed changes. Under these circumstances, we reserve the right to request a new submission in order that the safety aspects of the altered device can be given due consideration with regard to patient health and safety.
Clinical investigations also submitted to the FDA or other regulatory authorities
Manufacturers should clearly indicate whether the UK and non-UK protocols are the same. If not, the areas of difference should be referenced and an explanation of the reasons for the differences provided. It is recognised that the objectives of a clinical investigation which is also being carried out in a country or countries outside the UK, may be wider than those required by the UK MDR or EU MDR.
Changes to protocol requested by other regulatory authorities should be copied to the MHRA for information. Manufacturers should indicate whether the changes instigated by the non-UK regulatory authority will also be made to the UK protocol.
In-house manufactured medical devices
A ‘health institution’ is defined in Article 2(36) as meaning an organisation the primary purpose of which is the care or treatment of patients or the promotion of public health.
MDCG guidance notes that according to recitals 29 and 30 of the IVDR and MDR, health institutions include hospitals as well as institutions, such as laboratories and public health institutes that support the health care system and/or address patient needs, but which do not treat or care for patients directly.
The concept of health institution does not cover establishments primarily claiming to pursue health interests or healthy lifestyles, such as gyms, spas, wellness and fitness centres. The recognition as a health institution can also depend on national legislation and could thus differ between Member States.
Currently, the UK has no national provisions elaborating on what is or is not a health institution in Northern Ireland.
Under EU MDR Article 5(5), the notification requirements for clinical investigations will not apply to devices which are manufactured and used only within health institutions in Northern Ireland, provided the following conditions are met:
-
the devices are not transferred to another legal entity,
-
manufacture and use of the devices occur under appropriate quality management systems
-
the health institution justifies in its documentation that the target patient group’s specific needs cannot be met, or cannot be met at the appropriate level of performance by an equivalent device available on the market,
-
the health institution provides information upon request on the use of such devices to its competent authority, which shall include a justification of their manufacturing, modification and use
-
the health institution draws up a declaration which it shall make publicly available, including:
-
- the name and address of the manufacturing health institution
-
- the details necessary to identify the devices
-
- a declaration that the devices meet the general safety and performance requirements set out in Annex I to this Regulation and, where applicable, information on which requirements are not fully met with a reasoned justification therefore
-
the health institution draws up documentation that makes it possible to have an understanding of the manufacturing facility, the manufacturing process, the design and performance data of the devices, including the intended purpose, and that is sufficiently detailed to enable the competent authority to ascertain that the general safety and performance requirements set out in Annex I to this Regulation are met
-
the health institution takes all necessary measures to ensure that all devices are manufactured in accordance with the documentation referred to in point (f), and
- the health institution reviews experience gained from clinical use of the devices and takes all necessary corrective actions.
However, EU MDR Article 5(2) requires that “A device shall meet the GSPRs set out in Annex I which apply to it, taking into account its intended purpose.” Article 5(5) specifies that the need to meet relevant GSPRs still applies to devices/studies which are exempt from clinical investigation application requirements under the health institution exemption. Therefore, you must meet the requirements of both EU MDR Article 5(2) and 5(5) to qualify for in-house exemption.
Off-label use
If a clinician uses a UKCA or CE marked device for a new, off-label purpose that is unsupported by the manufacturer, then the clinician and the relevant healthcare establishment may take on the responsibilities of ‘the manufacturer’ if they see and intend a commercial application, and must therefore fulfil all the requirements of a manufacturer as set out in the UK MDR and EU MDR, including notification of a clinical investigation to the MHRA. They may also take on liability with reference to the device being used off-label.
You can find out more about the regulation of the off-label use of medical devices.
Research tools
It is the MHRA’s general opinion that a device being used on humans for research purposes, where there is no intended medical purpose for the device, could be a research tool. However, if a manufacturer sees and intends a medical application in the results generated from testing a device, then the device is no longer a research tool, but falls within the definition of a medical device.
Whether the regulations apply to a device will depend on the intended purpose foreseen by the device manufacturer. If a proposed clinical study includes investigating use of a device for a medical purpose, then such a study is likely to fall within the remit of the UK MDR or EU MDR and require notification to the MHRA as a clinical investigation.
We strongly recommend that manufacturers contact us for guidance on whether the regulations will apply before undertaking a study of this nature.
You should tell us:
- who has manufactured the device
- who is conducting the proposed study
- what the intended purpose of the device is
- what the intended purpose of the proposed study is
- whether any medical application is foreseen for the device
- a copy of the study protocol, where possible
Humanitarian use of non-UKCA or non-CE marked devices
The use of individual non-UKCA or non-CE marked devices may be authorised by the MHRA on humanitarian grounds, provided that the MHRA is satisfied that this would be in the interests of the patient and the protection of health. In such cases, the device may not be used until an application requesting such use has been made by the manufacturer and due authorisation has been given by the MHRA. Our authorisation applies only to the use of the individual device for a named individual within the UK. Failure to comply with these requirements is a criminal offence.
You must apply for humanitarian use of a non-UKCA or non-CE marked device.
Other factors to consider when planning a clinical investigation
Number of devices
In assessing risks to health or safety, one of the areas that will be particularly considered by the MHRA is the proposed number of devices to be included within a clinical investigation. The number must be sufficient to demonstrate performance satisfactorily and to reveal significant risks to patients’ health and safety. At the same time the number should not be so great as to place at risk more patients than necessary at a time when third party assessment of device-related risks has not been carried out
The number should:
- reflect the aims of the investigation
- take into account the perceived risk of the device
- comply with relevant medical devices standards where appropriate
You can find out more about statistical considerations for clinical investigations of medical devices.
Duration
The duration of a clinical investigation of a medical device should allow the demonstration of performance over a period of time sufficient to represent a realistic test of the device. It should allow time for the identification and risk assessment of any associated unacceptable adverse incidents over that period, allowing conclusions to be drawn as to the likely performance in the longer term.
It is not feasible or desirable to perform a clinical investigation lasting the projected lifespan of many devices. Indeed, it is recognised that for a number of devices (for example, orthopaedic implants and vascular stents) the majority of associated adverse incidents may not become clinically obvious for a number of years and that the clinical investigation in question will only demonstrate major short term safety problems.
The duration of a clinical investigation and follow-up period must be in line with relevant medical device standards where appropriate.
Post-market clinical follow-up
Article 61 of the EU MDR require manufacturers to actively update their clinical evaluation with data obtained from post-market surveillance.
It is intended that long-term safety problems be identified either under Medical Devices Vigilance or through a means of specifically designed post-market clinical studies, either extending the pre-market clinical investigation, by studying a relevant and identified cohort of patients over a defined period of time or through means of a specifically designed registry.
Where post-market clinical follow-up (PMCF) is not deemed necessary, this must be duly justified and documented.
In general, devices should follow a post-market clinical follow-up when one or more of the following criteria are identified:
- innovation, where the design of the device, the material, the principles of operation, the technology or the medical indication is new
- severity of the disease
- sensitive target population
- risky anatomical location
- well-known risks associated with a similar marketed device
- well-known risks identified from the literature
- identification of an acceptable risk during pre-market clinical evaluation, which should be monitored in a longer term or through a larger population
- identification of emerging risks in similar products
- obvious discrepancy between the pre-market follow-up windows and the expected life of the product
In Northern Ireland, you must notify the MHRA of all clinical investigations involving CE marked devices that also involve procedures additional to the normal conditions of use of the device, that are also invasive or burdensome. The notification should be made at least 30 days before the study commences. We will acknowledge the notification. If we have any concerns about the proposed study we will write to you.
Your notification submission will need to include:
- a completed application form
- investigators brochure
- clinical investigation plan
- signed statement
- research ethics committee opinion
- proof of insurance cover or indemnification of subjects
- patient information sheet and informed consent documentation
- arrangement to ensure protection and confidentiality of personal data
- details of the technical documentation (risk analysis, test reports) kept available
All notifications should be submitted using the application process on the Integrated Research Application System (IRAS) portal.
You can find out more in the guidance on clinical investigations for medical devices.
Type of investigation
The majority of clinical investigations of medical devices will not include a control group. The decision as to whether a control group is necessary however, will depend on the aims of the investigation. For some devices it would only be possible to demonstrate claims adequately by comparison with a separate or untreated group.
If control groups are necessary these should be randomised and prospective, except in exceptional and justifiable circumstances. Pivotal or confirmatory studies should have a control where clinically relevant and appropriate to do so. For all studies, lack of a control group should be justified.
End points
Care should be taken in choosing endpoints to ensure that this will support the stated aims and objectives of the clinical investigation under normal conditions of use. Methods of supporting the demonstration of these endpoints should, as far as possible, be objective. For example, derived from the results of diagnostic or in vitro diagnostic tests, rather than be a subjective factor, such as the severity of symptoms.
You can find out more about statistical considerations for clinical investigations of medical devices.
Labelling
All devices intended for clinical investigation must bear the wording ‘exclusively for clinical investigation’ as stated in Annex I, chapter III, 23.2(q) of the EU MDR.
This wording may cause confusion to clinical staff as it may be understood, mistakenly, that the clinical investigation being referred to is of a patient rather than the device. The MHRA recommended that manufacturers draw this requirement to the attention of all clinical investigators, ensuring that the meaning of this wording is clearly understood by all staff using or coming into contact with the device being investigated and that the device under investigation is segregated, where possible, from any similar devices in routine use.
If a device under clinical investigation has been UKCA or CE marked for another purpose, explanatory labelling to this effect should be attached to the device under investigation.
Research ethics committee opinions
For all clinical investigations of devices falling within the scope of the EU MDR, a relevant research ethics committee (REC) opinion is required as set out in EU MDR Article 62.3.
This opinion may be obtained in parallel with the MHRA notification. If the REC opinion is not provided at the time the application is made, it should be forwarded to us as soon as it becomes available.
No clinical investigation of an unmarked device should be started until both the relevant REC opinion and the MHRA have raised no grounds for objection. The MHRA does not accept approvals from independent ethics committees. Manufacturers must seek the opinion of a REC within the UK Health Departments’ Research Ethics Service. REC approval is required from just one REC, irrespective of the number of centres participating in the clinical investigation.
The Research Ethics Service can advise you on how to apply for a REC opinion. You can email the Health Research Authority (HRA) at queries@hra.nhs.uk. You should make it clear in your email if the investigation involves a non-UKCA or non-CE marked medical device.
If the MHRA raises no grounds for objection to the investigation in question proceeding, the investigation may only start once REC approval has been granted and a copy of the REC approval letter is sent by the manufacturer to the MHRA.
It may be helpful for us to liaise with the relevant ethics committee concerning notifications. It can also be helpful for the MHRA to send the committee a copy of the final decision for information purposes.
HRA approval and confirmed management permission
Each individual site in the clinical investigation must have confirmed management permission.
NHS organisations in England provide this by confirming that they have the capacity and capability to take part in the study.
In Northern Ireland, Scotland and Wales, NHS organisations provide a letter of NHS Permission.
Non-NHS organisations should confirm their management permission and receive a favourable Site-Specific Assessment from the REC.
The clinical investigation must not begin in the UK until you have received the relevant confirmation for that individual site. In addition, clinical investigations must not commence in any NHS site in England until they have also received HRA Approval.
You can get more detailed guidance from the HRA. You can also email the HRA at queries@hra.nhs.uk, making it clear that the investigation involves a non-UKCA or CE marked medical device.
After you have made your submission
When the MHRA has received your submission, we will follow our assessment process to decide whether to approve your clinical investigation.
You can find out more about what we do, and when, in our guidance on approving clinical investigations.
When you need to communicate with the MHRA
Making modifications to the investigation
All proposed changes to the clinical investigation must be notified to the MHRA, however only those considered to be substantial will require authorisation. Substantial modifications must not be implemented until we have confirmed authorisation.
Depending on whether we consult experts, we will issue a decision within 38 or 45 calendar days.
Substantial modifications
Substantial modifications are changes that are likely to have a substantial impact on the safety, health or rights of the participants or on the robustness or reliability of the clinical data generated by the investigation and include:
- changes to the medical device under investigation
- changes to the design or methodology of the clinical investigation, or to background information
- changes to the procedures undertaken by participants
- changes to the risk benefit assessment for the study
- significant changes to study documentation such as clinical investigation plan, investigator’s brochure, participant information sheets, consent forms, letters to GPs or other clinicians, information sheets for relatives or carers
- appointment of a new chief investigator
- inclusion of a new trial site (not listed in the original application)
- appointment of a new principal investigator at a trial site
- temporary halt of a study to protect participants from harm, and the planned restart of a study following a temporary halt
- a change to the definition of the end of the study
- extension of the study beyond the period specified in the application form
- any other significant change to the protocol
- changes requested by an ethics committee
Non-substantial modifications
For non-substantial modifications you only need to notify us to ensure our records are up to date. If upon review of the proposed non-substantial modification, we consider it to be in the substantial category, we will inform the sponsor, and the proposed modification should not take place until an MHRA authorisation is received.
These are changes that are unlikely to have a substantial impact on the safety, health or rights of the participants or on the robustness or reliability of the clinical data generated by the investigation and include:
- a change of sponsors or sponsors’ legal representative
- a change to the insurance or indemnity arrangements for the study
- minor changes to the protocol or other study documentation, for example, correcting typographical errors, updating contact points, minor clarifications
- changes to the chief investigator’s research team
- changes to the research team at particular trial sites (other than appointment of a new principal investigator)
- changes in funding arrangements
- minor changes in the documentation used by the research team for recording study data
- changes in the logistical arrangements for storing or transporting samples
Final clinical investigation report
Manufacturers are required to notify the MHRA when a clinical investigation comes to an end as stated in Article 77 of EU MDR.
You should provide us with a copy of the final clinical investigation report within 1 year of the end of the clinical investigation.
Early termination or temporary halt
Sponsors are required to notify the MHRA of the temporary halt or early termination of a clinical investigation and provide a justification within 15 days, or 24 hours if the decision was taken on safety grounds.
The clinical investigation report and summary must be provided within 3 months for studies that are temporarily halted or terminated early.
Adverse events involving devices undergoing clinical investigation
Sponsors are required to fully record all adverse events and report events occurring in all participating centres to the MHRA if there has been:
-
any serious adverse event that has a causal relationship with the investigational device, the comparator or the investigation procedure or where such causal relationship is reasonably possible
-
any device deficiency that might have led to a serious adverse event if appropriate action had not been taken, intervention had not occurred, or circumstances had been less fortunate
-
any new findings in relation to any event referred to in points (a) and (b).
These should be provided using the MDCG 2020-10/2 Rev.1 SAE reporting table.
You should submit a serious adverse event reporting form and the completed template form to the MHRA by using the MORE portal.
A ‘serious adverse event’ is any adverse event that led to any of the following:
a) death
b) serious deterioration in the health of the subject, that may have resulted in any of the following:
-
life-threatening illness or injury
-
permanent impairment of a body structure or a body function
-
hospitalisation or prolongation of patient hospitalisation
c) medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or a body function
d) chronic disease
e) foetal distress, foetal death or a congenital physical or mental impairment or birth defect
You can find out more in the guidance on clinical investigations for medical devices.
Quarterly summary reports
As a condition of MHRA approval for a clinical investigation, in addition to reporting individual serious adverse events as detailed above, you must send us quarterly summary reports providing an update on the latest overall safety profile for the investigation.
If the study is UK only, we expect the first QSR to be submitted one quarter after the first participant has been treated.
For studies that also have EU and global sites please provide a QSR one quarter after MHRA approval of the UK study. The report should detail the overall safety profile for ALL study sites.
To provide these summaries, use the guidance and template. This template is for devices only but you should also use it for device-related reporting on any combined studies. Do not include detail on any investigational medicinal product (IMP) under investigation.
Submit your quarterly summary reports directly through the MORE portal.
Study deviations
Manufactures must notify the MHRA of all deviations to the study as soon as they have been made aware of them. Details about the nature of the deviation, when it occurred, where it occurred, and any proposed corrective and preventative actions should be provided.
You should use the MHRA protocol deviation tracker when reporting deviations and keep this as a live document so that new deviations can be added. This enables both the sponsor and MHRA to have a complete overview each time it is submitted. You should send the completed spreadsheet to the MHRA by emailing info@mhra.gov.uk.
If the MHRA considers the regulations have not been met
Where there are grounds for considering that the requirements of the regulations are not met, we can:
- withdraw the no objection letter and revoke authorisation
- suspend or terminate the clinical investigation
- require the sponsor to modify any aspect of the clinical investigation
If such action is deemed necessary, the MHRA will write to the manufacturer or sponsor to communicate the decision and agree any further actions that may be necessary.
UKCA and CE-marking
Please inform MHRA in the event that the medical device under investigation is UKCA or CE marked. We request that at the same time the MHRA are also provided with a summary report of the clinical data from this clinical investigation that was used to support the UKCA or CE mark.
If the UKCA or CE marking covers the purpose under investigation, further modifications to the study documentation can be made without notification to the MHRA.
Further information
If you have any questions about the clinical investigation procedure you can email info@mhra.gov.uk.