Understanding haemoglobinopathies
Updated 11 July 2025

© Crown copyright 2025
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/sickle-cell-and-thalassaemia-screening-handbook/understanding-haemoglobinopathies
All healthcare professionals involved in the screening programme for sickle cell and thalassaemia (SCT) are advised to keep their knowledge updated; and an e-learning resource is provided to support practitioners.
1. Normal haemoglobin
Haemoglobin (Hb) is the substance within red blood cells which carries oxygen around the body[footnote 1].
Normal haemoglobin is made up of different globin (polypeptide) chains with heme molecules containing iron. The globin chains combine to make different types of haemoglobin. The structure of each globin chain in haemoglobin is genetically determined.
Normal haemoglobin (see illustration below) is called haemoglobin A and consists of:
- 2 alpha (α) globin chains
- 2 beta (β) globin chains
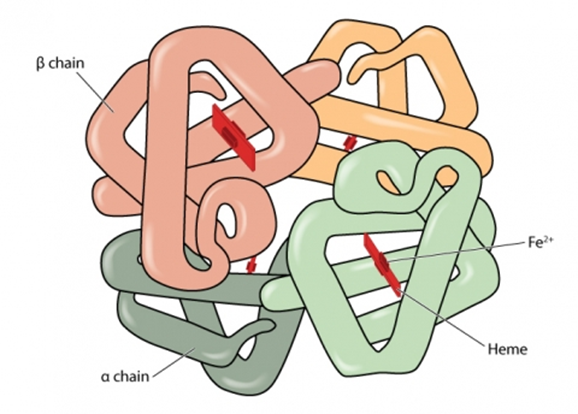
Adult red blood cells normally contain the following haemoglobin chain combinations:
- haemoglobin A (α2β2) >95%
- haemoglobin A2 (α2δ2) 2% to 3.4%
- fetal haemoglobin F (α2γ2) <1%
Laboratory tests can quantify normal haemoglobin and identify variants by their different characteristics.
2. Haemoglobinopathies: an overview
Haemoglobinopathies are a group of recessively inherited genetic conditions affecting the haemoglobin component of blood. They are caused by a genetic change (mutation) in the haemoglobin[footnote 2] [footnote 3]. More than 1,000 mutations[footnote 4] have been identified that result in either haemoglobin variants or thalassaemias.
Most of these unusual genes are clinically insignificant. However, there is a genetic relevance to some haemoglobinopathies which, when combined with other variants or thalassaemias, may cause a significant clinical condition resulting in illness and potential death.
The most significant haemoglobinopathies result in either a change in the structure and quality of the haemoglobin or a reduction in the quantity of haemoglobin produced.
Haemoglobinopathies are:
- not gender (x) linked
- more prevalent in some parts of the world
Sickle cell disease is most common in West Africa and India. Thalassaemia major is more common in Asia and Mediterranean countries.
The likelihood of a person being a carrier of a haemoglobinopathy depends on ancestry. The type of mutation varies between ethnic groups.
It is possible to inherit mutations in both alpha and beta globin genes at the same time.
It is also possible (although rare) for an individual to have a ‘de novo’ haemoglobin mutation. This is a genetic mutation that is not directly inherited from parents but is present only in that individual.
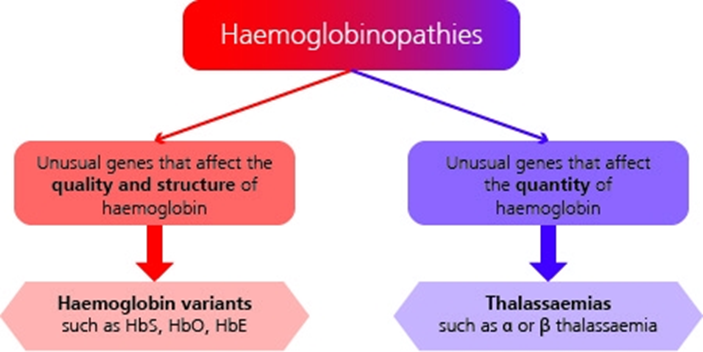
See illustration above for summary of haemoglobinopathy categories. See the NHS SCT Screening Programme adult carrier information leaflets for more information.
2.1 Change in structure and quality of haemoglobin
Haemoglobinopathies, where the mutation results in a change to the structure and quality of haemoglobin, are known as haemoglobin variants. The most important of these is sickle cell, Hb S. Other haemoglobin variants which have a genetic and possibly clinical significance (in combination with another haemoglobinopathy) are:
- Hb C
- Hb D
- Hb OArab
- Hb Lepore
- Hb E
Haemoglobin E (Hb E) is technically a haemoglobin variant. However, Hb E also interacts with beta thalassaemia to cause a significant clinical condition, so it can also be classified in the ‘thalassaemias’ category.
2.2 Reduction in quantity of haemoglobin
The thalassaemias is the name for a group of related conditions where the amount of haemoglobin that the body produces is reduced, and this impacts on its oxygen carrying capacity. These usually affect either the alpha or beta globin chain. The most significant of these are beta (β) thalassaemia, delta beta (δβ) thalassaemia and alpha (α) thalassaemia.
3. Inheritance of haemoglobinopathies
The genes for haemoglobin production are inherited from both parents. Please refer to the inheritance risk table for further details.
Haemoglobin disorders such as sickle cell disease or beta thalassaemia major are recessively inherited. If one unusual beta chain gene is inherited from one parent, the individual will be a carrier of the condition but will not be affected. This is sometimes called having a trait.
Carriers of haemoglobin variants are healthy and are unaware of their status unless screened. A carrier of a haemoglobin variant will usually have approximately:
- 50 to 65% normal haemoglobin A
- 30 to 45% unusual haemoglobin (for example Hb S, Hb C or Hb D)
- a small amount of haemoglobin A2 and F
If 2 unusual beta chain genes are inherited, one from each parent, the individual will have a haemoglobin disorder. The most common clinically significant conditions are beta thalassaemia major and sickle cell disease.
It is also possible to inherit a benign haemoglobin disorder, where the individual has 2 unusual haemoglobin genes but does not require treatment for a clinically significant condition. However, these conditions are genetically relevant.
For more information about haemoglobinopathies, please refer to the:
- haemoglobin carrier states in Appendix 1
- benign haemoglobin disorders in Appendix 2
If both parents carry a significant haemoglobinopathy, the baby is at risk of inheriting a haemoglobin disorder. There is a:
- 1 in 4 (25%) chance of being completely unaffected
- 2 in 4 (50%) chance of being a carrier
- 1 in 4 (25%) chance of inheriting the condition
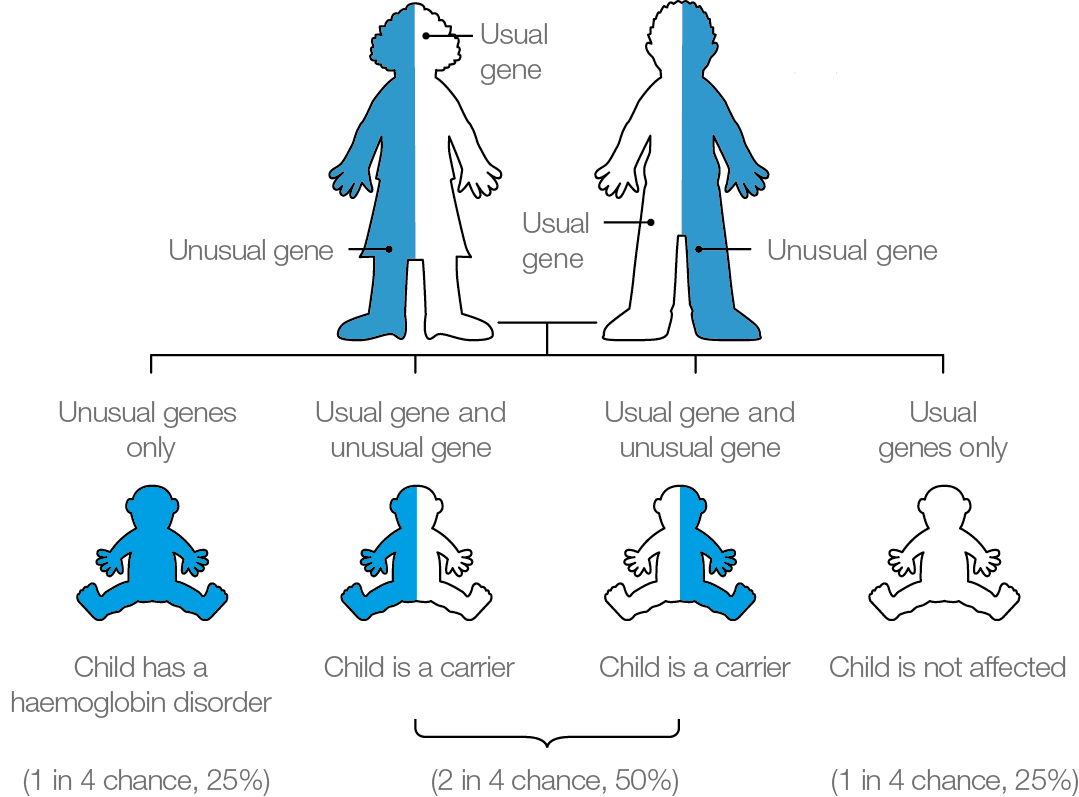
It is important parents are aware that the risks of inheriting a haemoglobin disorder are the same in every pregnancy (see diagram above).
4. Sickle cell disease
4.1 Sickle cell carriers
Sickle cell carriers have inherited one normal haemoglobin A gene and one sickle haemoglobin gene.
These individuals should be cautioned to be careful in situations where there may be a reduction in the supply of oxygen, for example when having a general anaesthetic or during activities such as deep-sea diving or mountain climbing. However, they do not have a condition which requires treatment.
See the NHS SCT Screening Programme leaflet for sickle cell carriers.
4.2 Sickle cell disease
Sickle cell disease is a recessively inherited genetic condition of haemoglobin[footnote 5]. It occurs when both parents pass unusual haemoglobin genes to their baby.
Sickle cell disease most commonly affects people of West African, Caribbean, Middle Eastern and Indian ancestry. However, it can affect anyone from any population.
The most common types of sickle cell disease seen in England are:
- haemoglobin SC disease (Hb SC)
- Hb S/beta (β0/β+) thalassaemia
- sickle cell anaemia (Hb SS)
In an individual with sickle cell disease, the red blood cell becomes misshapen and rigid, resembling the shape of a sickle, when the haemoglobin is de-oxygenated (releases the oxygen to the organs). This process is called sickling and causes a wide range of clinical complications.

Normal red blood cell

Sickled red blood cell
4.3 Prevalence of sickle cell disease in England
Sickle cell disease affects around 1,000 pregnancies per year and there are between 260 to 350 babies born with a sickle condition each year. Between April 2007 and the end of March 2017 there were 3,206 babies born with sickle cell disease in England.
The National Haemoglobinopathy Register (NHR) has a record of more than 11,000 individuals with sickle cell disease in England. However, registration is voluntary, so the actual number is likely to be higher.
4.4 Clinical characteristics of sickle cell disease
Normal red blood cells move freely in the circulation and have a life span of 120 days. De-oxygenated sickled red blood cells can get stuck and cause blockages in capillaries (small blood vessels) and have a shortened life span of approximately 20 to 30 days, sometimes less. These blockages are known as vaso-occlusive episodes and are sometimes described as a painful crisis.
A sickle cell crisis can be triggered by:
- sudden changes in body temperature
- dehydration
- shortage of oxygen
- infection
‘Sickling’ can result in:
- intense pain
- severe anaemia
- tissue damage
- infections
- strokes, particularly in individuals with Hb SS
- shortened life expectancy
Appendix 3 has further details of the clinical consequences of sickle cell disease based on specific genotypes.
5. Management of individuals with sickle cell disease
Sickle cell disease requires specialist consultant haematologist or paediatrician management. Early diagnosis is vital and screening for sickle cell disease is incorporated in the NHS newborn blood spot (NBS) screening programme in England. Children who are diagnosed with sickle cell disease at birth should be referred for specialist care by 3 months of age.
Standards have been developed for appropriate care of children and adults with sickle cell disease.
Management of individuals with sickle cell disease incorporates:
- regular specialist outpatient reviews and easy, direct access to specialist medical care when unwell
- prophylactic antibiotics and regular immunisations to prevent infections
- consideration of hydroxycarbamide for children with sickle cell anaemia (Hb SS) over 9 months of age or if the individual has significant and frequent symptoms
- care with administering general anaesthesia which should only be undertaken when full medical support is available (this also applies to dental treatment)
- blood transfusions for relevant complications, for example stroke or severe life-threatening anaemia
- annual transcranial doppler scans to ascertain increased risk of stroke for children and adolescents with sickle cell anaemia (Hb SS) and Sβ0 thalassaemia, up to the age of 16 years[footnote 6]
Sickle cell disease can be cured by bone marrow or stem cell transplant, but the genetic profile of the individual does not change. One potential side effect of undergoing this procedure is infertility.
Much of the care required by individuals with sickle cell disease is preventative and supportive care. Families and individuals need specialist support to understand the condition and be proactive in their personal care, to recognise potential problems and try to prevent or minimise the effects of a sickle cell crisis. Preventative care includes:
- eating a balanced, healthy diet
- maintaining a good fluid intake
- keeping warm in cold conditions
- complying with recommended immunisations and prophylactic antibiotics
- administering simple analgesia at the start of a sickling episode
- seeking urgent medical care when required
6. Thalassaemias
Thalassaemias are usually recessively inherited genetic conditions which affect the quantity of haemoglobin produced. A change occurs in the genetic code responsible for production of either the alpha or beta globin chains that are present in normal haemoglobin A. The 4 main thalassaemia conditions that have clinical significance are:
- alpha thalassaemia major, which is clinically significant to the fetus and mother
- haemoglobin H disease (alpha thalassaemia 3 gene deletion), which is clinically significant after birth
- beta thalassaemia major, which is clinically significant after birth
- beta thalassaemia intermedia, which has variable clinical significance after birth
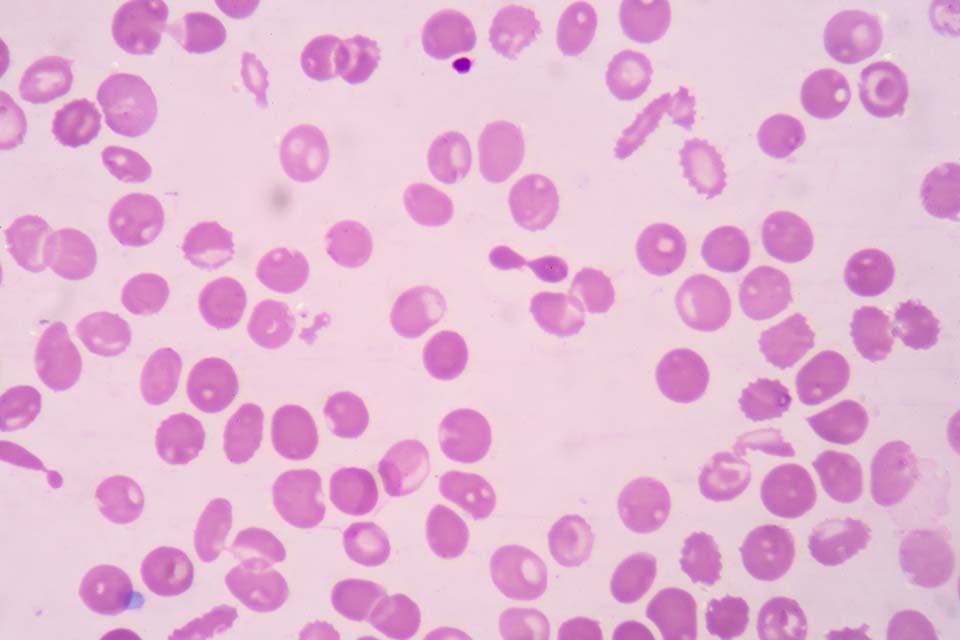
Thalassaemic red blood cells
Further details on the clinical impact of thalassaemia conditions is in Appendix 4.
7. Alpha thalassaemia
Normal haemoglobin A has 2 alpha globin chains. The production of these alpha globin chains is controlled by 4 alpha globin genes and 2 genes are inherited from each parent. In alpha thalassaemia there is either reduced or absent production of alpha globin chains, caused by a defect or mutation in one or more of the alpha globin genes[footnote 7].
Alpha thalassaemia carrier status cannot be definitively diagnosed by routine laboratory screening methods, due to its complexity. It must be confirmed by DNA analysis.
Below is a graphic illustration of the alpha globin chains (2), and the full complement of alpha globin genes, (4) that individuals with normal Hb A inherit. This is usually written αα/αα in medical literature.
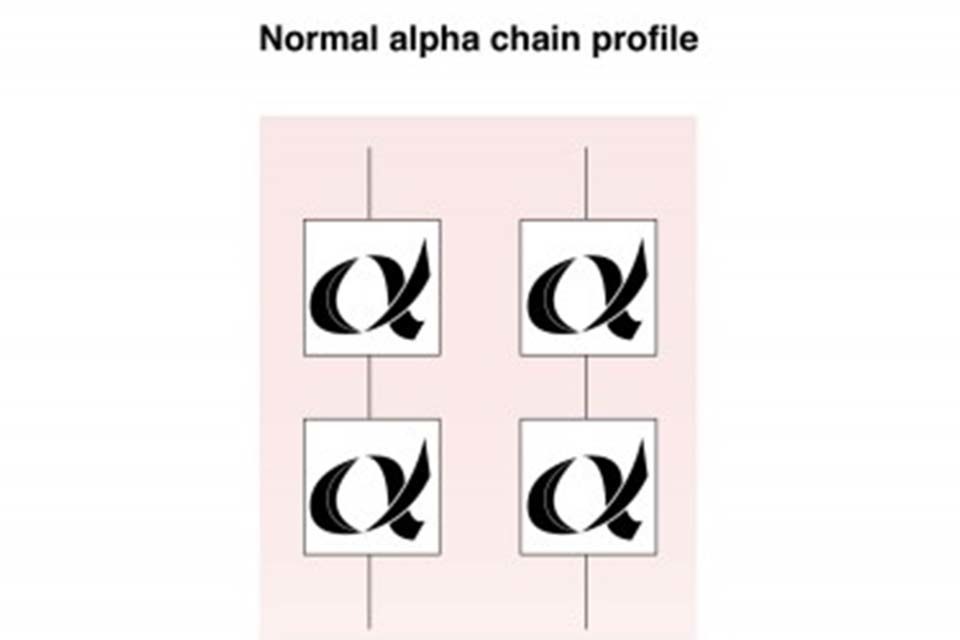
Structure of normal haemoglobin A alpha globin chains
7.1 Alpha plus (α+) thalassaemia carrier
Individuals with alpha plus thalassaemia have inherited either one or 2 faulty alpha globin genes (-α/αα) or (-α/-α). Although this can affect alpha globin chain production, there is usually minimal change to the haemoglobin level.
Alpha plus thalassaemia carrier status is not clinically significant but can be confused with iron deficiency anaemia in the antenatal period.
Alpha plus thalassaemia is most often seen in African, Caribbean, Indian, Pakistani, Bangladeshi and Middle Eastern populations.
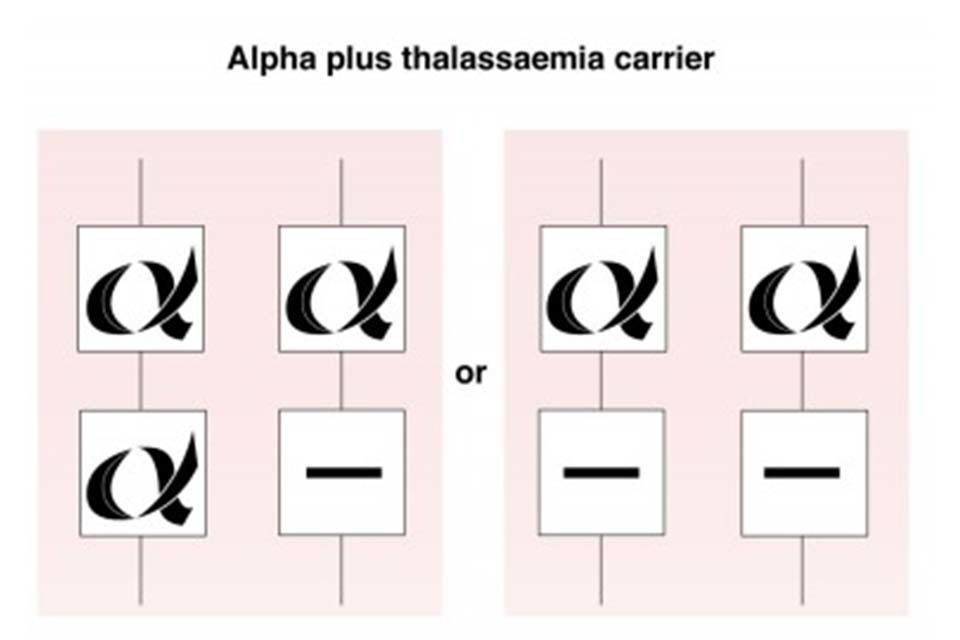
Alpha plus thalassaemia carrier
7.2 Alpha zero (α°) thalassaemia carrier
Alpha zero thalassaemia carriers have not inherited any alpha globin chain genes from one parent (- -/αα). The individual is generally healthy but there is a reduction in alpha globin chain production and they may have a mild anaemia with a mean cellular haemoglobin (MCH) that is usually less than 25pg. This can be confused with iron deficiency anaemia.
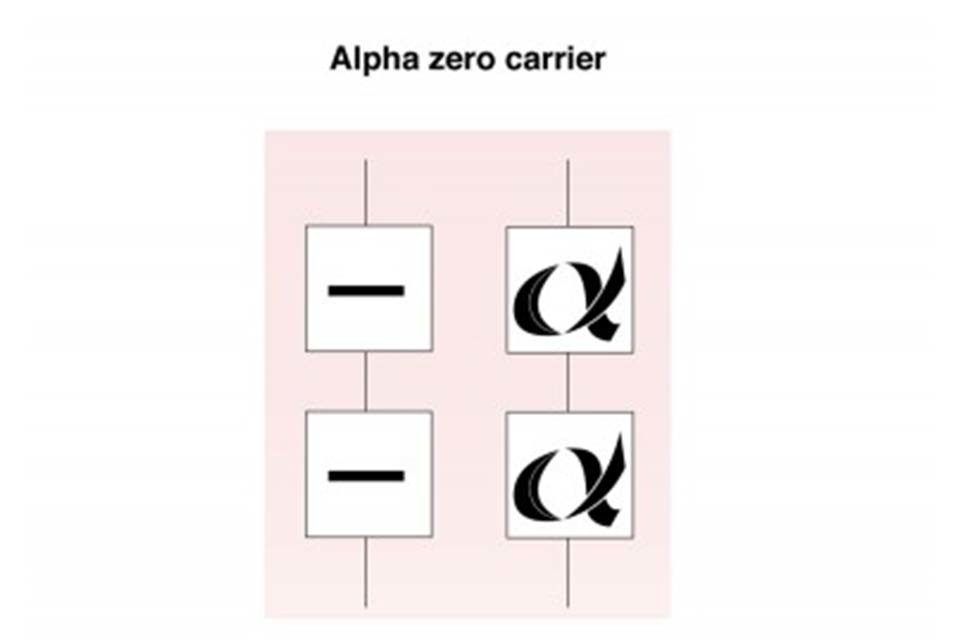
Alpha zero thalassaemia carrier
If a couple are both identified as potentially being alpha zero thalassaemia carriers, then their carrier status must be confirmed by DNA analysis as there is a 1 in 4 (25%) risk to all their children of inheriting Bart’s Hydrops Fetalis, also known as alpha thalassaemia major.
7.3 Haemoglobin H disease
An individual with haemoglobin H disease (- -/-α) has only one functioning alpha globin gene but usually retains the ability to produce sufficient haemoglobin for life. The haemoglobin level usually ranges from 70g/l to 100g/l, with a mean cellular haemoglobin (MCH) of 15pg to 25pg.
Haemoglobin H disease is a mild to moderate condition. It does not usually require treatment or lifelong blood transfusions, however short-term transfusions may occasionally be required during critical periods, such as pregnancy or illness or if the individual has an infection.
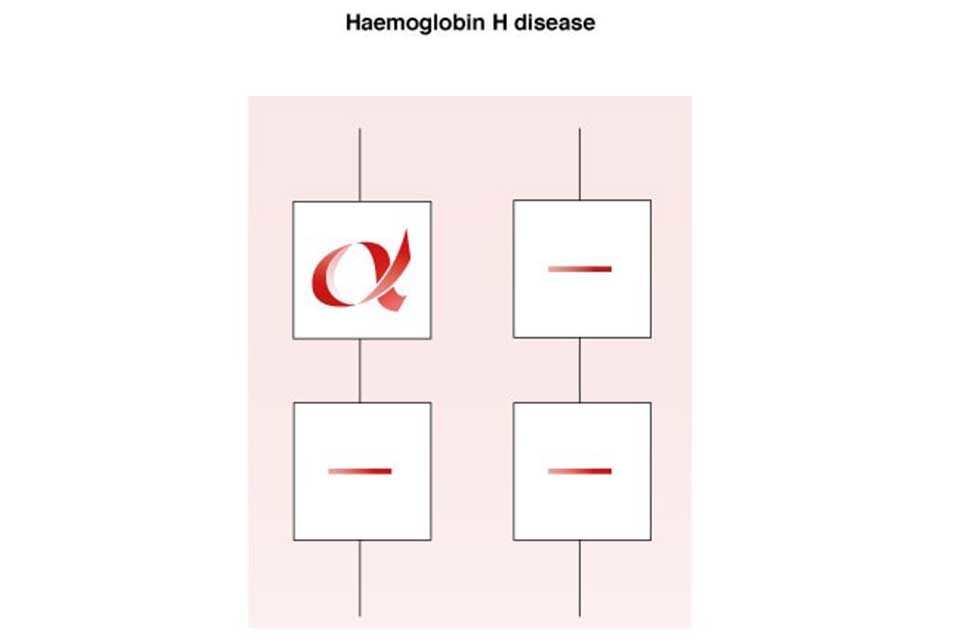
Haemoglobin H Disease
7.4 Alpha (α°) thalassaemia major
In alpha thalassaemia major, also known as Bart’s Hydrops Fetalis, there are no functioning alpha globin genes (- -/- -). As a result, no alpha globin chains are produced, which results in a severe life-threatening anaemia in the fetus and, without intervention, is incompatible with extra-uterine life.
The hydropic fetus can usually be diagnosed by ultrasound scan during the second trimester of pregnancy. Very occasionally, babies survive with intra-uterine transfusions. However, there is a high risk of significant disabilities after birth.
Implications for the mother during pregnancy with a fetus that has Bart’s Hydrops Fetalis include:
- pre-eclampsia
- ante-partum haemorrhage
- retained placenta
- possible 50% maternal mortality rate if alpha thalassaemia major in the fetus is undiagnosed
During antenatal screening it is important to diagnose if both parents are alpha zero (α°) thalassaemia carriers. Essential components for this are:
- accurate family origin information
- blood tests (analysis of full blood count)
- DNA analysis for confirmation of carrier status
The family origins at highest risk of alpha zero (α°) thalassaemia carrier status are from Taiwan, Laos, Vietnam, Malaysia, China, Hong Kong, Myanmar (Burma), Cyprus, Turkey, Sardinia, Greece, Singapore, Philippines, Thailand, Cambodia and Indonesia.
In the UK around 20 to 30 couples annually are identified as being at high risk of having a baby with alpha thalassaemia major.

Inheritance of alpha thalassaemia major
8. Beta thalassaemia
There are 2 main thalassaemia conditions depending on the genotype of the beta thalassaemia gene mutation inherited. They are:
- thalassaemia major
- thalassaemia intermedia
8.1 Beta thalassaemia carriers
A beta thalassaemia carrier:
-
has inherited 2 haemoglobin A genes, one of which is faulty and can usually be classified as one of the following:
- beta+ (beta plus) thalassaemia – where a reduced amount of Hb A is produced
- beta0 (beta zero) thalassaemia – where no Hb A is produced by that gene
-
has changes to the red blood cell indices:
- haemoglobin A2 is usually between 3.5% to 8%
- Hb F is greater than 1% but less than 10%
- MCH (mean cellular haemoglobin) is usually below 27pg
-
is not usually diagnosed at birth using routine newborn screening methods, due to the high level of fetal haemoglobin which masks the inheritance of a faulty haemoglobin A gene. Babies who may have inherited a beta thalassaemia gene should be tested when they are over 9 months of age to confirm their carrier status
A beta thalassaemia carrier may be misdiagnosed as having iron deficiency anaemia because the red blood cells are smaller and paler than usual, and there may be a lower than normal haemoglobin level. However, iron supplements must not be given to correct the apparent ‘anaemia’ caused by carrier status, as they are ineffective.
Further details are available in the leaflet Information for adult haemoglobinopathy carriers – you are a beta thalassaemia carrier.
8.2 Beta thalassaemia intermedia
Thalassaemia intermedia occurs in an individual when the beta globin chain production is significantly reduced but not completely absent. Clinical implications vary depending on the gene mutations inherited from both parents. The individual usually has a degree of anaemia, but the condition is not as severe as beta thalassaemia major.
The individual usually manages without regular blood transfusions but there may be splenomegaly and the requirement for occasional blood transfusions during pregnancy or illness. Regular monitoring by a specialist is recommended.
8.3 Beta thalassaemia major
Beta thalassaemia major is also called ‘Cooley’s Anaemia’ or ‘Mediterranean Anaemia’[footnote 8] and is most common in people of Pakistani, Cypriot, Italian, Greek, Indian, Bangladeshi, Chinese and other East/South East Asian ancestry.
In beta thalassaemia major there is severely reduced or absent production of the beta globin chains that make up normal adult haemoglobin, due to defective beta globin genes which are inherited from both parents. This results in severe, life-threatening anaemia which usually requires regular blood transfusions to sustain life.
8.4 Prevalence of thalassaemia conditions in England
It is estimated that thalassaemia major affects about 1 in 27,000 pregnancies. Although there is no specific newborn screening programme, using the current technology most babies with beta thalassaemia major are identified during the newborn screening process. There are approximately 20 to 30 babies born with beta thalassaemia major each year.
The NHR has a record of more than 1,200 individuals with thalassaemia conditions. As inclusion in the registry is voluntary, it is possible the total number is greater than this.
8.5 Clinical complications of untreated beta thalassaemia major
Clinical complications of untreated beta thalassaemia major include:
- failure to thrive in babies and young children
- lethargy and fatigue due to severe anaemia
- hypersplenism due to increased breakdown of red blood cells
- over activity of the bone marrow and bone deformity
- severe anaemia leading to early death
9. Management of individuals with beta thalassaemia major
Although beta thalassaemia major is not included in the NHS NBS Screening Programme, children with this condition are usually identified and should be referred for specialist care. Early diagnosis helps to monitor a child’s condition until blood transfusions are required. It also gives parents the opportunity to learn about the condition before complications arise.
The management of individuals with beta thalassaemia major aims to correct the severe anaemia and includes:
- blood transfusions every 3 to 5 weeks, usually starting from approximately 9 to 12 months of age
- iron chelation therapy to remove the excess iron (either orally or by subcutaneous injection)
- regular hospital appointments to monitor the condition
- splenectomy for hypersplenism
Supportive care is important for people affected by beta thalassaemia major. They:
- should be encouraged to avoid iron rich foods and have a daily vitamin C supplement
- may be suitable for bone marrow or stem cell transplant to cure their condition, although their genetic profile does not change
- may need psycho-social support in addition to medical care
There is a risk of early death unless patients adhere to the strict regime of blood transfusions and iron chelation.
Thalassaemia major can be cured by bone marrow or stem cell transplant, but the genetic profile of the individual does not change. One potential side effect of undergoing the procedure is infertility.
9.1 Clinical complications of treated beta thalassaemia major
If thalassaemia major is not well managed by transfusion therapy this could result in anaemia and over-activity of the bone marrow. However, complications are usually related to an excess of iron accumulated in the body due to the regular blood transfusions. This can result in:
- damage to the pituitary glands which could affect:
- growth and delay puberty
- insulin production, resulting in diabetes
- hypothyroidism
- cardiac impairment/failure
- liver damage
- lethargy and fatigue
- erectile dysfunction in men and amenorrhoea in women
- change in skin colour due to iron deposits
Individuals may also have:
- hypersplenism due to increased breakdown of red blood cells
- increased susceptibility to bacterial infections especially if the spleen has been removed
- Increased risk of blood borne infections due to regular blood transfusion therapy
10. Haemoglobin E/beta thalassaemia
Haemoglobin E/beta thalassaemia may result in a syndrome similar to beta thalassaemia major or beta thalassaemia intermedia. However, some individuals do not have any significant clinical symptoms.
As there is no way to predict the severity of the condition at birth, all babies should be referred to a specialist for assessment and follow-up.
11. References
-
Sickle Cell Information Centre (SCINFO) Tutorial: Normal Blood Cells ↩
-
Bain BJ (2006) Other significant haemoglobinopathies. Haemoglobinopathy Diagnosis, 2nd Edition Blackwall Publishing ↩
-
Weatherall DJ, Clegg JB (2001) Inherited haemoglobin disorders: an increasing global health problem. Bulletin of the World Health Organisation, 2001, 79 (8) (article contains prevalence data) ↩
-
Huisman THJ et al A database of Human Hemoglobin Variants and Thalassemias ↩
-
Eleftheriou A, Angastiniotis M About Sickle Cell Disorders. Thalassaemia International Federation (TIF) ↩
-
UK Forum on Haemoglobin Disorders (2016) Transcranial doppler scanning for children with sickle cell disease: standards and guidelines 2nd edition ↩
-
Eleftheriou A, Angastiniotis M About Alpha Thalassaemia Thalassaemia International Federation (TIF) ↩
-
Eleftheriou A, Angastiniotis M About Beta Thalassaemia Thalassaemia International Federation (TIF) ↩