Newborn screening
Published 24 November 2021
Applies to England

© Crown copyright 2021
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/sct-screening-handbook-for-newborn-laboratories/newborn-screening
Newborn sickle cell screening is offered for all babies born in England and the other UK countries at 5 days of age as part of the NHS newborn blood spot (NBS) screening programme. Additionally, babies up to 12 months of age who become the responsibility of the provider organisation must be offered screening if there is no documented evidence of a conclusive result for the conditions currently recommended by the UK National Screening Committee (UK NSC).
The NHS NBS screening programme offers screening for 9 conditions including sickle cell disease (SCD). Parents can choose to accept or decline screening for SCD separately from the other 8 conditions.
The objectives of newborn sickle cell screening are to:
-
improve infant health through prompt identification of babies born with conditions and timely transition into clinical care
-
achieve the lowest possible childhood death rate and to minimise childhood morbidity from sickle cell diseases
The linked antenatal and newborn NHS Sickle Cell and Thalassaemia (SCT) Screening Programme aims to:
- promote an appropriate level of understanding about screening for these genetically inherited conditions among professionals involved with the programme
- review the results from antenatal testing before, during and after the newborn test is offered, and to check that the results are congruent
- prepare parents for their baby’s screening result
It is essential that babies with these conditions are diagnosed reliably, that they are clearly reported as having a sickle cell disease and that the necessary clinical follow-up is arranged.
The analytical methods used by laboratories involved in newborn screening will detect most cases of β thalassaemia major and related conditions. See acceptable analytical protocols section. Alpha and β thalassaemia carriers are not identified.
The NBS screening programme has published standards for newborn screening, against which the screening services will be assessed.
1. Clinically significant haemoglobinopathies that must be detected
Current screening methods detect many haemoglobin (Hb) variants. Those for which there is evidence that early intervention is likely to be beneficial are specified as part of the national screening programme.
Clinically significant sickle cell disease includes:
- Sickle cell anaemia (Hb SS)
- Hb SC disease
- Hb S/βthalassaemiai
- Hb S/DPunjab
- Hb S/E
- Hb S/OArab
- Hb S/Vii
- Hb S/HPFHiii
i This is inclusive of Hb S/β+, Hb S/β0, Hb S/δβ, Hb S/γδβ, Hb S/εγδβ and Hb S/Lepore.
ii ‘V’ as denoted in Hb S/V in this publication has been in use since the first edition (2005) as a technical term to designate variants which are not S, C, DPunjab, E, and OArab. As there are more than 1,000 it is not practical to list them. This term is only used in the analytical/technical descriptions and has never been intended nor recommended for use in reports. The clinical advisor to the NHS Sickle Cell and Thalassaemia (SCT) screening programme has recommended that babies with the FSV pattern (fetal haemoglobin F, sickle haemoglobin S and unidentified haemoglobin variant V) are reported as “condition suspected” (status code 08). See the Reporting results section of handbook. These variants do not require full identification by the newborn first or second-line laboratories but must be referred for clinical assessment and passed into the diagnostic pathway where applicable. This also acts as a failsafe for screening laboratories where a baby with sickle cell disease has a coexisting alpha chain variant that may result in diagnostic confusion. See analytical results and status codes in Reporting newborn sickle cell results table.
iii In general HbS with hereditary persistence of fetal haemoglobin (Hb S/HPFH) is regarded as a milder condition than the other sickling conditions. Clinical follow-up is offered to distinguish it from other more significant abnormalities. It is not possible at birth to differentiate those conditions which produce only HbF and HbS on analysis; Hb SS, Hb S/β thalassaemia syndromes and Hb S/HPFH. For the purpose of this programme it is essential to detect and report all such cases as ‘results consistent with sickle cell disease’ without further detail to enable follow up and diagnostic testing.
Since there are many Hb ‘D’ variants with the same analytical characteristics (unless using more specific techniques such as tandem mass spectrometry (MSMS)) and characterisation of the variant may take time, it is recommended that all ‘D’ haemoglobins with the same analytical characteristics of DPunjab (also called DLos Angeles) are provisionally identified as this haemoglobin, the only clinically significant haemoglobin D variant. DNA analysis or mass spectrometry can then be used to confirm the diagnosis when clinically indicated.
2. Beta thalassaemia syndromes
Beta thalassaemia syndromes are another group of conditions that are detected by the screening programme where the patient will benefit from follow up. The UK NSC has agreed that, in line with other national screening programmes, clinically significant findings of conditions which are not part of the screening programme, but are detected by current screening methods, must always be reported to the relevant clinician to enable management of the consequences of such findings.
Little or no haemoglobin A (HbA) on newborn screening is clinically significant since it may indicate little or no β-chain synthesis. The policy of the screening programme is that in all cases where the apparent HbA concentration is 1.5% or less of the total haemoglobin the result must be reported as F only (or FE only if E is present) and followed up clinically. A review of the efficacy of this approach has been published [footnote 1] [footnote 2].
Newborn screening laboratories must report possible Hb E/β thalassaemia. This is because many of these children will become transfusion dependent or have thalassaemia intermedia. Note that homozygous HbE and Hb E/β0 thalassaemia will look identical on the initial screening test and will need to be differentiated in the diagnostic pathway.
3. Other carriers and ‘clinically benign’ haemoglobinopathies likely to be detected by screening
While the main purpose of this programme is to detect babies with sickle cell disease, most of the analytical procedures currently in use also detect homozygotes and compound heterozygotes, and carriers of the other common haemoglobin variants (C, DPunjab, E and OArab). Results of babies who are found to be homozygotes or compound heterozygotes for a common haemoglobin variant must be reported by the laboratory and follow-up counselling should be offered. These are listed below.
3.1 Conditions that are usually clinically benign
These are:
- Hb CC and C/β thalassaemia
- Hb DD and D/β thalassaemia
- Hb CD
- Hb CE
- Hb DE
- Hb EE
For MSMS, all samples with results outside the designated action values must be sent for second-line testing. See Action values for tandem mass spectrometry screening.
When using high performance liquid chromatography (HPLC), capillary electrophoresis (CE) or isoelectric focusing (IEF) a small number of other variants may be detected. These may not be immediately identifiable, and most will be benign. The number of neonates with one of these other variants is likely to be small. It is national policy that laboratories must not report variants other than S, C, DPunjab, E and OArab (this includes Hb Bart’s). However, laboratories must send certain specimens for second-line testing. These are:
- samples with 1.5% HbA or less
- samples with variants (peaks) more positively charged than HbA (eluting after HbA by HPLC and located to the right of HbA on CE)
This makes sure that:
- samples with little or no normal adult haemoglobin (HbA) have the result confirmed before reporting
- HbS (or one of the designated haemoglobins) is not missed even if it falls outside the predefined analytical windows
HbOArab, which has no defined analytical window, will still be detected. Using HPLC or CE and IEF there is a risk that newborn HbOArab carriers may not be positively identified. This is due to the difficulty in obtaining appropriate quality control material. Hb S/OArab compound heterozygotes will be detected and followed up clinically as per the recommendations for all cases with HbS and no HbA.
The Reporting results section of the handbook provides the recommended wording for reporting the results for these mainly clinically benign variants, either homozygous, compound heterozygous or carriers and uses the newborn status code 04 ‘Condition not suspected’.
For any other report, the recommended wording must state that ‘haemoglobins S, C, DPunjab, E and OArab have not been detected. Note that carriers of β thalassaemia and Hb Lepore are not detected by the techniques used for newborn screening’.
4. Sample requirements
Four good quality spots are necessary to complete the proper processing of the specimen. The demographics must be completed so the sample can be processed and to assist patient follow-up. See guidelines for newborn blood spot sampling.
Due to the nature of dried blood spots, there is deterioration of the blood sample from the time it is taken, as oxidation of the haemoglobin results in methaemoglobin formation. This degradation is likely to be greater at higher temperatures, but in normal circumstances it should not prevent analysis using the techniques described in Acceptable analytical protocols. In occasional cases where there is a delay in the card being sent to the laboratory, or if it is kept in unsuitable conditions, excessive oxidation may occur, rendering the sample unsatisfactory for analysis.
5. Pregnancies at high risk of a clinically significant haemoglobinopathy
Women known to be at high risk of having a baby with sickle cell disease or β thalassaemia major might wish to know the result for their child before the normal time for reporting the result from the blood spot. Local policies should be in place to have a newborn blood spot for HbS screening taken earlier if requested. Alternatively, a liquid capillary blood specimen (not cord blood) can be taken for analysis soon after birth. This is not currently part of the screening pathway and must be considered as an aspect of parental choice.
This blood specimen must be analysed in a specialist laboratory, which has expertise in haemoglobinopathy analysis in the newborn period. The laboratory must participate in an ISO 17043 accredited scheme for liquid newborn samples. It is the responsibility of the laboratory undertaking such an analysis to ensure that this result correlates with that from the newborn screening blood spot.
The fact that such a specimen has been taken should be noted by the midwife on the newborn screening blood spot card. The screening laboratory will undertake the routine screen as usual and this test will act as a ‘failsafe’ and quality check. It is imperative that the test result is given as soon as possible to decrease anxiety about the status of the child. Testing of the liquid specimen must be seen as a parallel test to the screening specimen and not a substitute.
6. Risks of transfusions containing red cells on screening results
The presence of transfused red cells in the neonate will interfere with the interpretation of the results from the haemoglobin analysis of the blood spot. This could possibly invalidate the results. A small number of babies are transfused in utero but for babies transfused post delivery it must be policy in all neonatal units to take a blood spot specimen for sickle cell screening before giving a transfusion. See guidelines for newborn blood spot sampling for requirements for these special circumstances.
Where a pre-transfusion specimen is not available, DNA extracted from the white cells in the blood spot is analysed, thus overcoming the complications caused by the presence of transfused red cells on the blood spot card. Kings College Hospital and Sheffield Children’s Hospital provide this service for England. This service does not eliminate the need to take a pretransfusion specimen.
The DNA test will detect the presence of the sickle globin gene. This test is able to differentiate between babies with:
- only the sickle gene present
- those with the sickle gene and another globin gene (either a normal beta gene or a beta gene with another variant)
- no sickle gene present
If the sickle gene is detected the baby must be referred for clinical follow up. This test does not confirm the identity of the non-sickle haemoglobin. Parents may want further standard haematology tests on their baby if they are known to be at risk of another haemoglobinopathy. This is not considered part of the newborn screening programme and should be initiated in a clinical setting. Testing using techniques other than DNA should not normally be undertaken until at least 4 months after the last transfusion.
7. Percentages of HbA in untransfused babies at different gestational ages
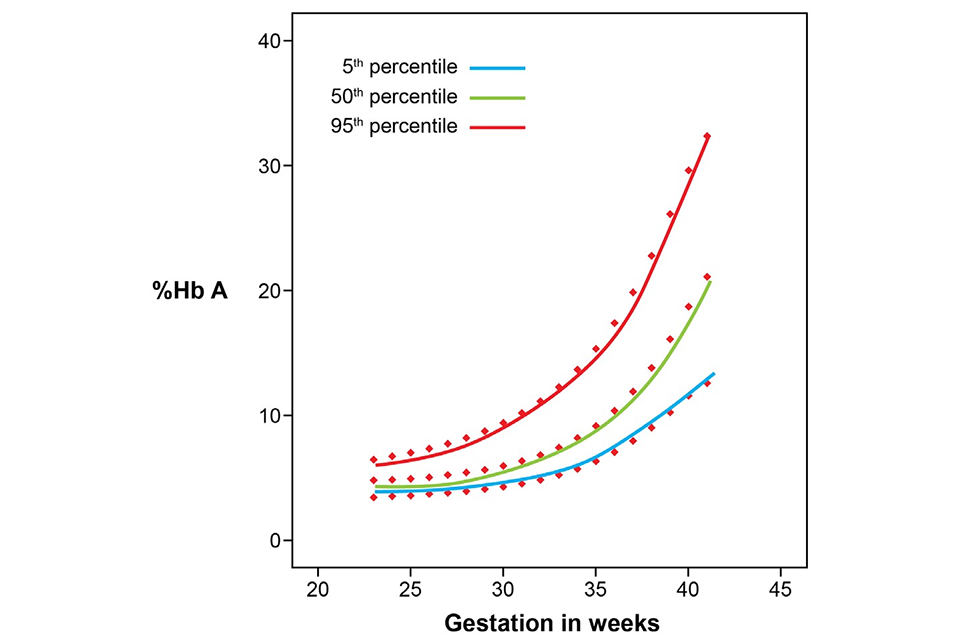
Figures 1 and 2 below are derived from data provided by the newborn screening laboratory of Birmingham Children’s Hospital, which used a BioRad VNBS analyser with valley-to-valley integration. The figures provide guidance about the expected levels of adult haemoglobin (HbA) in newborn babies and babies up to one year of age. The data on newborn babies is derived from 30,000 measurements and is presented as a percentile chart. The data on older babies from one month to one year is derived from 89 measurements and is therefore less statistically valid.
The purpose of the graphs in figures 1 and 2 is to provide guidance about the expected percentage of HbA that is seen in babies born from 23 weeks gestation up to one year of age. Increased percentages of HbA can be found following transfusion. The graphs may help to determine if the amount of HbA is appropriate for the age of the baby.
The primary way in which the transfused baby should be identified is the information provided by the healthcare professional who completes the blood spot card. However, this data field on the card is not always completed.
7.1 Figure 1: percentage of HbA in routine specimens taken from untransfused babies with gestations from 23 to 42 weeks
7.2 Figure 2: percentage of HbA in untransfused babies one month to one year old
The newborn screening laboratories can use the information provided in these graphs to reduce unnecessary referrals for DNA testing, especially in older babies. This is important as the failsafe screening test for sickle haemoglobin on transfused babies using DNA methods must only be used when a baby has received a transfusion and no pre-transfusion sample has been taken.
8. Sample analysis
Newborn screening for SCD using haemoglobin eluted from dried blood spots can be reliably undertaken using a primary screen to detect the different haemoglobin fractions present. In the case of suspected abnormality, a second-line test on the same specimen using a different scientific principle must be carried out to validate the initial findings.
It is important to note that unequivocal identification of haemoglobin variants can only be achieved by either protein sequence analysis by mass spectrometry or analysis of DNA. Occasionally the presumptive identification of a haemoglobin variant using screening methods is incorrect due to comigration of variants. Screening is not a diagnostic service and no screening programme has a sensitivity and specificity of 100%. However, haemoglobins S, C, DPunjab, E and OArab should be detected reliably.
-
Streetly A, Latinovic R, Henthorn J, Daniel Y, Dormandy E, Darbyshire P, Mantio D, Fraser L, Farrar L, Will A, Tetlow L. ‘Newborn blood spot results: predictive value of screen positive test for thalassaemia major’. Journal of Medical Screening 2013: volume 24, issue 4, pages 183 to 187 ↩
-
Daniel Y, Henthorn J. Reliability of the current newborn screening action value for beta thalassaemia disease detection in England: A prospective study. Journal of Medical Screening 2019: 26(2):67-79 https://doi.org/10.1177/0969141318797373 ↩