Guidance on pharmacovigilance procedures
Updated 8 January 2025

© Crown copyright 2025
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/guidance-on-pharmacovigilance-procedures/guidance-on-pharmacovigilance-procedures
1. General Approach to the operation of pharmacovigilance
The MHRA retains responsibility for Pharmacovigilance across the UK.
There are some different requirements for products placed on the market in the UK with respect to Great Britain and Northern Ireland. Great Britain is England, Wales and Scotland. For products authorised for sale or supply in Northern Ireland EU pharmacovigilance requirements will continue to apply in addition to UK requirements as indicated in this guidance.
For medicines which are authorised nationally in the UK, you as a Marketing Authorisation Holder (MAH), will be required to submit pharmacovigilance data to the MHRA, according to UK requirements, including:
- UK and non-UK Individual Case Safety Reports (ICSRs)
- Periodic Safety Update Reports (PSURs)
- Risk Management Plans (RMPs)
- Post-Authorisation Safety Studies (PASS) protocols and final study reports
These will be assessed taking into account all relevant information and decisions will be made reflecting UK clinical practice to best support patient safety in the UK.
The Good Vigilance Practices (GVP) modules will remain in force but a guidance note on the exceptions and modifications to the EU guidance on good vigilance practices has been published and you should refer to these guidelines when preparing your submissions
In addition to these requirements, you are reminded that you are obliged to notify us of any information that you consider might influence the evaluation of the benefits and risks of your product as soon as is reasonably practicable after you become aware of it, including where the use of the product is restricted in any country. This is particularly important where any new information may potentially impact the clinical management of patients, including where proactive communications to healthcare professionals may be required.
You are also reminded that you are responsible for ensuring that the information for your product is kept up to date with current scientific knowledge. This includes checking the MHRA website, the European Medicines Agency (EMA) and Head of Medicines Agencies (HMA) websites for the outcome of procedures which affect the product information and, unless advised otherwise, you should implement the updates to the product information in the UK via the appropriate variation application.
More information on specific areas is given as follows:
2. Actions for submitting and receiving ICSRs
We require submission of all UK (including Northern Ireland) ICSRs (serious and non-serious) and serious ICSRs from other countries via the MHRA Gateway and/or ICSR Submissions portal.
Further information on sending and receiving information on adverse drug reactions (ADRs) to the UK can be found at Adverse drug reactions (ADRs)
You can register on the MHRA-Gateway and/or ICSR Submissions portal to enable configuration to your systems.
For products placed on the market in Northern Ireland you should continue to submit ICSRs according to EU requirements to the Eudravigilance database including all serious reports from the UK and other countries and non-serious reports that occur in the EEA or in Northern Ireland. These cases should be identified by using the country code “XI” in the field primary source country for regulatory purposes. Organisations have the option to use either the country code “XI” or “GB” as the first two characters of the worldwide case ID and the safety report ID.
For UK cases that relate to Northern Ireland which were initially submitted before 1 January 2021, the worldwide case ID should not be changed, however, in line with GVP module VI guidance when sending follow-up reports organisations can change the safety report case ID if needed.
The country code “GB” should be used for all reportable SUSARs occurring in the UK. Although it is possible for organisations to use the country code “XI” for SUSAR reporting, it will not be a requirement as reporting requirements between Northern Ireland and the rest of UK for clinical trial cases will not differ.
3. Signal detection
Your signal detection systems need to enable you to meet your requirements for cumulative signal detection across all available data sources. The MHRA does not require you to conduct signal detection against our own database, as we will make relevant UK data available for inclusion in your systems.
The MHRA will carry out assessment of signals and issue decisions for signals identified by the MHRA as well as those highlighted internationally.
For all products authorised in the UK you are obliged to notify us of new information arising from any data source (except our database) which impact on the marketing authorisation. This includes standalone signal notifications submitted by you to the EMA that are relevant to your UK products as well as signals raised by the EMA. However, you do not need to inform us of signals once they are on the PRAC agenda.
Details of how to notify the MHRA of this new information is provided below.
You should inform us in the following circumstances:
3.1 Emerging safety issues
You are obliged to notify the MHRA of emerging safety issues within 3 working days after establishing that a signal or a safety issue from any source which meets the definition of an emerging safety issue (see GVP-Module IX Signal Management)
You should notify the MHRA at signalmanagement@mhra.gov.uk
3.2 Signals for UK authorised products
You should inform us of signals only once they have been validated. Validation of your signal should include a thorough analysis of ICSR data available and be complemented by an assessment of other relevant information including whether the risk is addressed in other UK-authorised products.
You should notify us of these validated signals by the following routes:
Variation of the MA
Where you conclude, based on your assessment that there is a new or changed risk which requires a change to the product information and /or Risk Management Plan (RMP) you should submit a variation application. In this case a separate standalone signal notification is not required as the proposed changes and supportive evidence will be assessed within the variation procedure by the licensing authority.
Where a variation to address a signal has been submitted to the EU and affects products authorised in the UK, you should also submit the variation to the MHRA. Where relevant, the variation can be submitted via the reliance route and, in this case, the EU assessment report, CHMP opinion and EC decision (where relevant) should be submitted once available. In these circumstances MHRA will consider the EU outcome in the context of the UK clinical situation. Normally, we would expect a reliance procedure to be submitted once the CHMP opinion is available, however, where new information might influence the evaluation of the benefits and risks, is likely to impact clinical management of patients and/or require proactive communications (e.g., via a DHPC) you are obliged to inform us as soon as possible. In these circumstances you should submit the variation to the MHRA in parallel to the EU irrespective of whether the reliance route is to be used. Where the reliance procedure is to be used the MHRA will generally not conclude the variation until there is an EU decision (either CHMP opinion or EC decision, where relevant).
Where you have submitted a variation to any regulatory authority outside of the UK and the change impacts products authorised in the UK you should make an assessment as to whether the variation should also be submitted to us.
Inclusion in the Periodic Safety Update Report (PSUR)
If a PSUR is due to be submitted within 6 months of the completion of your assessment of the signal, the signal should be reported in the PSUR, and a separate standalone signal notification is not required unless we advise you otherwise. However, if the PSUR includes a signal that corresponds to an important risk (see GVP Annex 1), you should notify us separately at signalmanagement@mhra.gov.uk at the time of submission of the PSUR. This also applies if you have been asked to assess a signal by another regulatory authority outside of the UK. Refuted signals need only be reported in PSURs.
Standalone signal notification
Where the above circumstances do not apply further analysis can be sought from us where a validated signal cannot be refuted nor confirmed as a new or changed risk by your assessment. In this case you should submit the signal as a standalone signal notification at signalmanagement@mhra.gov.uk
Refuted Signals
Refuted signals need only be reported in PSURs
If you are notifying us of a signal in circumstances other than those described above you should provide an explanation as to why you are doing so and how the validated signal impacts products authorised in the UK, together with your proposed action.
3.3 EU-led signal assessments
For signals on the Pharmacovigilance Risk Assessment Committee (PRAC) agenda we request that you keep us updated with any further information available to you, including the outcome of the assessment and, where relevant, your proposed actions regarding your UK product. We request that any PRAC signal assessment reports available to you are shared with us once the PRAC recommendation is available. Signal assessment reports should be submitted to signalmanagement@mhra.gov.uk.
3.4 Products authorised for use in Northern Ireland
For products authorised in Northern Ireland, in addition to the above requirements, where you detect a new signal when monitoring EudraVigilance you should validate the signal and inform the EMA.
You are additionally required to report to the EMA those safety signals that are considered to meet the definition of an emerging safety issue (see GVP-Module IX Signal Management).
4. Risk Management Plans (RMPs)
Submission of a new Risk Management Plan (RMP) or an update to an existing RMP may be required at any time during the product’s lifecycle. RMPs should continue to follow the EU template GVP module V Risk Management Systems Where your product has an approved RMP you should consider whether changes to the RMP are required whenever new data becomes available. Situations where an updated RMP is required include new safety concerns or where there is a new or significant change in the existing pharmacovigilance or additional risk minimisation activities.
If your product does not have an approved RMP (products authorised prior to July 2012), circumstances where you should introduce one include where there are significant changes to the MA or where there are new or changed safety concerns that affect the balance of the benefits and risk of the product.
We continue to accept approved versions of the EU RMP but where there is GB/UK specific information this can be provided in a GB/UK specific annex using this format. We encourage you to use this format where appropriate but where you have already had an RMP-annex approved in a different format there is no need to convert to this format.
Where you are applying for a new MA or other procedure which necessitates a GB/UK specific RMP you can either submit a GB/UK RMP or use the approved EU RMP, if one is available, with a specific GB/UK annex. The GB/UK-specific RMP annex should be attached to the corresponding EU RMP, placing the GB/UK-specific annex first and the EU RMP second in a single document. In cases where the differences between GB/UK and EU RMPs are extensive and affect the majority of sections in the RMP, a standalone GB/UK RMP should be prepared using the EU RMP template.
Where a new or updated RMP needs to be submitted outside of a regulatory procedure it should be submitted to the MHRA using the appropriate variation procedure. This includes circumstances where you have an approved GB/UK RMP but wish to apply to update it in line with the approved EU RMP.
If the RMP needs to be updated because of a PSUR assessment it should be submitted to the MHRA via a variation procedure. The MHRA PSUR portal cannot accept RMPs.
4.1 Additional Risk Minimisation Measures (aRMMs)
The MHRA is responsible for the oversight of additional risk minimisation measures (aRMMs) required for products authorised in the UK. Additional risk minimisation measures should be submitted to the MHRA for agreement before implementation in the UK. Where the aRMMs are in the form of educational materials these need to be submitted to us together with details about their distribution to RMPallocation@mhra.gov.uk. Further guidance on educational materials can be found at Guidance on educational materials. Where there is a requirement for implementation of additional risk minimisation measures with respect to Northern Ireland (for example, for a centrally authorised product) the case for any differences in approach for the rest of the UK should be discussed with the MHRA.
5. Periodic Safety Update Reports (PSURs)
5.1 Format and content of PSURs
The format and content of the PSUR is set out GVP module VI and the associated exceptions document. The expectation is that where a PSUR is required to be submitted to both the MHRA and EU, the same PSUR will be submitted to both authorities. However, where the MHRA has made a specific request for information or where there is UK-specific information relevant to the benefit/ risk assessment, this should be included in a specific annex. There is no template for this annex as the content will depend on the specific issues to be covered but if you have concerns about the information to include, please contact us for further advice at vigilanceservice@mhra.gov.uk.
5.2 Submission of PSURs
PSURs for UK products should continue to be submitted according to the EU Reference Date (EURD) list.
Unless the marketing authorisation specifies differently, PSURs for actives/combinations not currently on the EURD list, should be submitted at least six monthly during the first 2 years following placing on the market, once a year for the following 2 years and every 3 years after that. The exception to this is where the product is authorised under Article 10(I) or 10a, in these circumstances PSURs are not required unless there are specific requirements in the MA.
PSURs for Great Britain-only MAs should be submitted to us via our PSUR portal (please see below). PSURs for UK-wide or Northern Ireland-only MAs should be submitted to the EU PSUR repository. We no longer require these PSURs to be submitted separately to us provided that we have access to them from the EU PSUR repository. Further information on submission requirements is provided below:
PSURs for Great Britain-only MAs
We may develop our own submission requirements for PSURs for products authorised via a Great Britain-only MA and publish a list of UK reference dates. However, in the meantime, you should follow the EU reference date (EURD) list for PSUR submissions to the MHRA and submit according to the specified dates.
For products authorised in Great Britain before the MA is granted in the EU you will be advised of PSUR submission requirements at the time of authorisation. There will be the facility to request alignment of submission with the EURD list later, depending on when the product is authorised in the EU. Where this applies you should contact us to discuss the specific circumstances.
You should submit your PSUR to us via our PSUR submission portal. Detailed requirements for how to submit to our portal can be found at making submissions to the MHRA but PSURs can be submitted to the portal in PDF or word format or as part of a zip file format. You should submit your PSUR via the procedure number on the portal. This will be the same as the procedure number on the EURD list without the ‘USA’ e.g.PS/12345678/123456. Submissions are not required as part of the eCTD lifecycle in the UK.
Submission requirements for PSURs for products authorised in Northern Ireland
PSURs for products authorised in Northern Ireland, should be submitted to the EMA via the EU PSUR repository. This includes PSURs for products subject to UK-wide authorisations (i.e., covering both GB and NI) and NI-only MAs. These PSURs are also required by MHRA, however, as we have access to the EU PSUR repository, to avoid dual submission where you have a UK MA covering NI, you are no longer required to submit the PSUR to the MHRA. This is provided your PSUR has been submitted to the EU PSUR repository and we have access to it from there. We do, however, ask you to e-mail at safetyprojects@mhra.gov.uk and inform us when you submit your PSUR to the EU PSUR Repository.
In some circumstances, we may request submission of the PSUR via our portal where we consider it necessary, and you should follow the instructions for submission to our portal where required (please see further details above).
Informal PSUSA follow-up procedure
Where your PSUR is subject to an informal PSUSA follow-up procedure (please refer to the CMDh website for further details on this procedure) please inform us by e-mail at safetyprojects@mhra.gov.uk when the procedure is initiated. You should continue to submit the information regarding the procedure via the route specific for this procedure (i.e., via CESP not the EU PSUR repository or MHRA submission portal). Outcomes should be implemented via the same type of variation procedure as in the EU and where the MHRA have additional requirements we will inform you.
5.3 Fees
A fee of £890 will be payable for PSURs which are required to be submitted to our portal and are for actives/ combinations which are currently listed on the EURD (or UK reference date list). There will be a reduction to £445 for each PSUR where more than one PSUR is involved in the procedure and submitted to the MHRA. Please note you will need a purchase order (PO) ready when you submit. No further fee will be payable for the amendment of the product information as a result of the UK assessment which should be made by a Type IA variation.
5.4 Assessment and outcomes
PSURs subject to the EU single assessment process
Where the EU assessment results in an amendment to the product information, please notify us of the PRAC recommendation as soon as possible and submit a copy of the CMDh Decision or CHMP opinion as soon as available. This information should be submitted by e-mail to safetyprojects@mhra.gov.uk. This will allow us to take the EU recommendation into account in our assessment and avoid any unnecessary divergence. We will inform MAHs directly if, following our assessment, we have any additional requirements to the EU. Provided information regarding the EU assessment is submitted to us in a timely manner this will be communicated before or shortly after (within 14 calendar days) the publication of the EU outcome, otherwise the EU outcome should be implemented by the same type of procedure as in the EU. A delay in providing the necessary information to the MHRA may result in additional requests being made outside this timeframe.
Where the outcome for the corresponding centrally authorised product is implemented as part of the EC Decision without a separate variation, unless you are advised otherwise, a Type IAIN variation should be submitted to implement the same outcome for the Great Britain-only MA.
MHRA will not routinely issue PSUR assessment reports in addition to those circulated by the EU.
PSURs subject to UK only assessment
Where there are issues to address, we will contact MAHs directly.
6. Post Authorisation Safety Studies (PASS)
6.1 PASS protocols and results
PASS Protocols
The following advice refers to non-interventional studies, for PASS which fall under the definition of a clinical trial please refer to the relevant guidance.
Where the study is imposed i.e. is a specific obligation or a condition of the UK MA (category 1 and 2 studies), the draft protocol should be submitted to the MHRA prior to the start of the study. Where the MA applies in Northern Ireland, the draft study protocol should also be submitted to the Pharmacovigilance Risk Assessment Committee (PRAC). The exception to this is a study that is only to be conducted in the UK at the request of the MHRA, in which case the protocol should only be submitted to the MHRA. For studies that are a condition of a GB MA or an UK MA, where the study is to be conducted in the UK at the request of the MHRA, the MHRA will assess the protocol and contact you within 60 days.
Where the PASS is imposed, any significant amendments to the draft protocol should also be submitted to the MHRA and where the MA applies in Northern Ireland, additionally to the PRAC (unless the study is only being conducted in the UK at the request of the MHRA). The definition of significant amendment is in line with that provided in Good Vigilance Practice Module VIII.
There is no specific requirement to submit draft protocols for PASS which are voluntary (not a condition of the MA) including non-imposed studies that are requested as part of the RMP (category 3 studies). However, we may request submission where we consider that an assessment is required, in this case the protocol should be submitted as Type II (standard) variation.
PASS Results
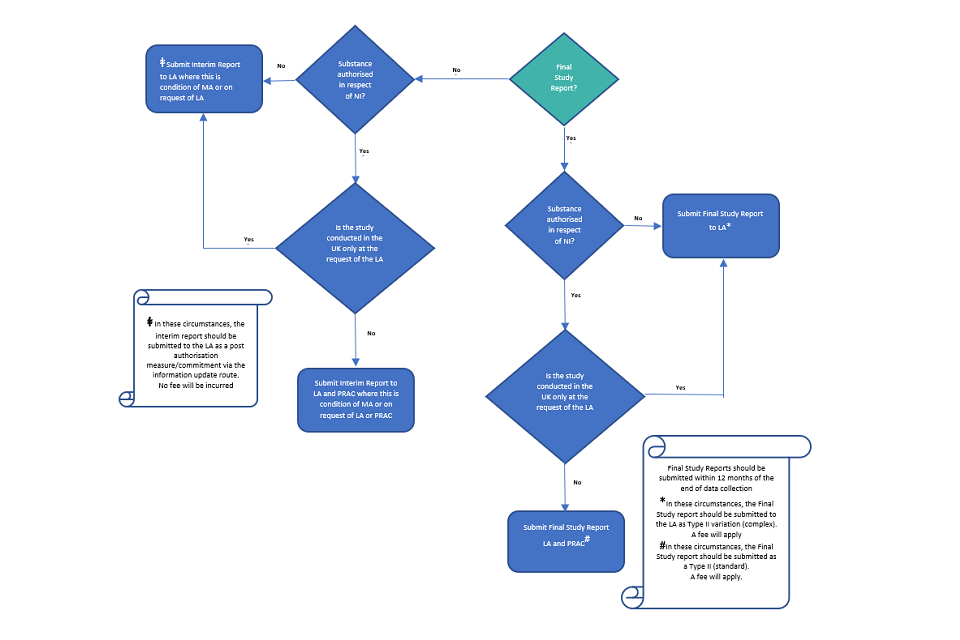
Final study reports for all non-interventional PASS (either voluntary or imposed) that involve collection of safety data from patients/healthcare professionals should be submitted to the MHRA.
The final study report should be submitted within 12-months of the end of data collection.
Interim reports for PASS which are a condition of the UK MA should also be submitted to the MHRA.
Where the MA applies in Northern Ireland, interim reports for PASS which are a condition of the MA and all final study reports for non-interventional PASS, should also be submitted to the PRAC (unless the study was only conducted in the UK and not at the request of PRAC)
Interim reports for non-imposed studies are generally not required, but where we require interim reports for these PASS, we will inform you.
6.1.2 Submission and fees
Imposed PASS
- Protocol and Final Study reports
Protocol and final study reports should be submitted to the MHRA when available.
Where protocols and final study reports are also submitted to the PRAC, the MHRA will take the EU decision into consideration and the relevant EU documentation (assessment report, CHMP opinion etc.) should be provided as soon as available. A reduced fee of £734 will apply and to facilitate submission, protocols and final study reports should be submitted to the MHRA using a Type II (standard) variation procedure (classification C.I.13).
Where the PASS is to be conducted only in the UK or relates to a product authorised only in Great Britain and the protocol and final study report have not been submitted to the PRAC, the protocol and final study reports should be submitted to the MHRA. A fee of £8,309 will apply in this case and to facilitate submission, protocols and final study reports which fall into this category should be submitted via a Type II (complex) variation procedure (classification C.I.13).
- Significant amendments and interim study reports
Significant amendments to study protocols and interim study reports for imposed PASS should be submitted to the MHRA as post authorisation measures/ commitments via the information update route and will not incur a fee. It should be clearly stated in the cover letter that the amendment or interim report relates to an imposed PASS. These submissions will be dealt with as post authorisation measures (PAMs) (please see section 10).
Non-imposed PASS
This includes PASS requested in RMPs (category 3 studies). Where draft protocols are requested, they should be submitted via the Type II (standard) variation route. Final study reports for non-imposed PASS should always be submitted to us, usually via a Type II (standard) variation procedure but where the final study report is also submitted to the PRAC, the same route of submission can be used for the MHRA (either Type II or as a. post authorisation measure) (also see section 10 on post-authorisation measures). The type II variation route should always be used where the product information is affected.
Where protocols and final study reports are also submitted to the PRAC, the MHRA will take the EU decision into consideration and the relevant EU documentation (assessment report, CHMP opinion etc.) should be included in the submission to us.
Where interim reports are required for non-imposed PASS these should be submitted to us as post authorisation measures/ commitments via the information update route and will not incur a fee (please see section 10)
6.1.3 Updates to product information to reflect study results
Where the results of any PASS study affect the product information, the amendments should be submitted via a type II variation. The results and amendments can be submitted as part of the same variation provided the appropriate fee is paid (£734 where the study results and amendments are also submitted to the PRAC, otherwise £ 8,309).
The submission requirements for PASS are summarised in Figs 1 and 2.
7. Safety Referrals
UK products continue to be part of Union referral procedures in respect of Northern Ireland. The scientific opinion and Commission or CMDh Decision will include UK products in respect of Northern Ireland. You should implement the outcome via the relevant variation procedure where your MA covers Northern Ireland.
Where you hold a Great Britain-only MA, and the outcome of an EU referral is to be reflected in your MA you should submit a Type II variation application together with all relevant documentation regarding the referral. This will be assessed by the MHRA, taking into account the EU decision, to determine the necessary actions.
You are reminded that where new information comes to light which might influence the evaluation of the benefits and risks of your product you are obliged to inform us as soon as is reasonably practical. We therefore expect to be informed of the initiation of a referral procedure that affects products authorised in the UK and kept informed of the relevant data and assessment. Please inform us at signalmanagement@mhra.gov.uk.
8. Major Safety Reviews
Where there are concerns regarding a medicine or class of medicines that are authorised in the UK, the MHRA may conduct a major safety review to review the available data and consider what regulatory action may be needed. We will initiate a review independently of the EU in circumstances where the nature and magnitude of the safety concern has implications for the positive benefit/risk balance of the medicine or class of medicines including where the initial review of the safety concern suggests that significant restrictions to use may be needed such as introduction of a contraindication, restriction to the authorised indication, or amendment to the authorised posology or where the safety concern potentially has significant public health implications or is of particular public concern.
Fig 1: Submission Requirements - Study Protocol or Substantial Protocol Amendment for Non-Interventional PASS
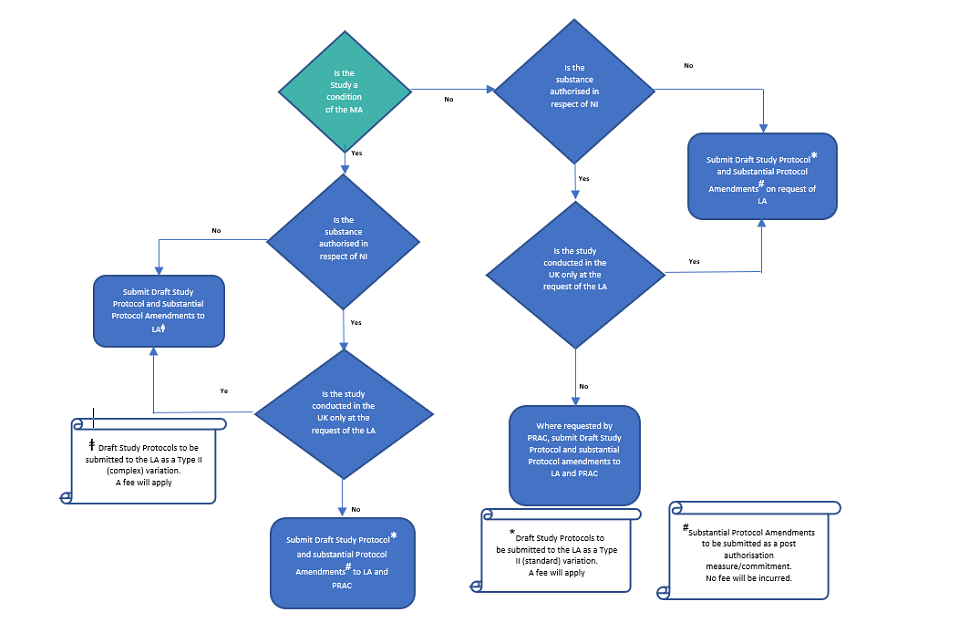
Fig 2: Submission Requirements - Final Study Report or Interim Study Results for Non-Interventional PASS
In these circumstances we will publicly announce the initiation of the review, outlining the reasons for the review, the list of affected active substances and products, and the timescales for the review.
Where you hold a MA for an affected product, you will be notified of the start of and reasons for the review. You will also be provided with a list of questions that should be addressed by all MAHs along with the deadline by which the requested information should be submitted.
In the first instance, this correspondence will be done via the Qualified Person for Pharmacovigilance (QPPV) but a different or additional contact for future correspondence can be nominated. The outcome of the review will be published. Where the recommendations include proposals for regulatory action the details of the measures to be taken including any changes to the product information will be published.
A major safety review will incur the following fees for assessment:
- £51,286 where one or two active ingredients or combinations of active ingredients are included
- £59,595, where three active ingredients, or combinations of active ingredients, are included
- £67,904, where four active ingredients, or combinations of active ingredients, are included
- £76,213, where five or more active ingredients, or combinations of active ingredients, are included
Where the review relates to 2 or more authorisations, the fee will be divided by the number of authorisations forming part of the review and you will pay that reduced fee for each relevant authorisation you hold.
9. Other MHRA safety reviews
We will also conduct safety reviews for UK products in circumstances where a major safety review does not apply, this may be in response to a new safety signal or in order to support effective risk minimisation. In this case we will contact MAHs directly with the outcome of the review and any wording to be implemented. We will publish a public assessment report relating to our safety reviews wherever appropriate. There will be no fee for our review, but normal variation fees will apply for the implementation of any wording to the product information.
Where the review impacts on products authorised via the MR/DC procedure we will not make a direct request for a variation, however, our expectation is that you will review the new safety information and take forward the issue as necessary within the MR/DC procedure, in line with your obligation to keep the MA updated in line with current scientific knowledge. As stated in CMDh guidance it is not possible to implement the outcome of a MHRA safety review to a product in the MR/DC procedure via a TIB procedure and any update should be submitted via a Type II variation application CMDh minutes SEPT 2021 . We will provide you with the assessment report which you can use to support your MR/DCP application, but you can supplement this with your own information as appropriate. We will also contact the RMS, or CMDh where several RMS are involved, and inform them of our review and outcomes. CMDh has advised member states that they can contact us directly where they require further information CMDh minutes OCT 2021
If your product is authorised via the MR/DC procedure you cannot implement any update on a purely UK basis, however, there is the option to withdraw from the MR/DC procedure and have a UK national MA if you choose Handling of MR.DR procedures.
10. Post-authorisation Measures (PAMs)
Post-authorisation measures and commitments including specific obligations (SOBs), Annex II conditions, additional pharmacovigilance activities in the RMP (MEA), legally binding measures (LEG) or recommendations (REC) in place on 1 January 2021 will remain in place for converted EU MAs until considered fulfilled. For MAs granted via the reliance route after 1 January 2021 all post-authorisation measures and commitments agreed for the EU MA will also apply.
Where an obligation or condition or MEA. falls under the definition of a non-interventional PASS please follow the guidance for submission outlined in sections 6.
10.1 Submission of information in relation to PAMs
Specific obligations (SOB)
Specific obligations (SOB), imposed on marketing authorisations granted under exceptional circumstances or on conditional authorisations, form the basis of the annual re-assessment or annual renewal. SOBs may also be additional pharmacovigilance activity and included in the RMP (category 2 studies). SOBs are binding conditions and any modification with regards to the description should be submitted within an appropriate procedure, either the annual re-assessment, annual renewal or via a variation. Interim results which do not affect the product information, which are submitted outside the annual re-assessment or renewal can be submitted via the PAM submission route as described below. Final results leading to fulfillment of a SOB should be submitted within the appropriate procedure (annual re-assessment, the annual renewal, or a variation application).
Annex II conditions (ANX)
Annex II conditions, imposed obligations to conduct post-authorisation measures, are considered to be key to the benefit risk of the product and may also be additional Pharmacovigilance activity and included in the RMP (category 1 studies). As they are binding conditions any modification with regards to the description should be submitted via a variation procedure. Interim results which do not affect the product information can be submitted via the PAM submission route as described below. Final results leading to fulfillment of an Annex II condition should be submitted via a variation application.
Additional pharmacovigilance activity in the risk-management plan (MEA)
Additional pharmacovigilance activities in the RMP (category 3 studies) are studies required to investigate a safety concern. Any changes to additional pharmacovigilance activities agreed within the RMP should be submitted via the appropriate variation procedure to amend the RMP. Information not impacting on the product information or description/ or due date of the measure itself can be submitted via the PAM route as described below. Final results leading to fulfillment of a MEA should be submitted via a variation application.
Legally binding measures (LEG)
Where a LEG procedure is initiated in the EU, please inform us by e-mail safetyprojects@mhra.gov.uk once the procedure has been initiated. Subsequent information should be submitted via the PAM route (see below).
Recommendations (REC)
Recommendations are non-binding to the MA but where they are made information concerning the REC can be submitted as a PAM other than where there is an impact of the product information.
Where any information regarding a PAM affects the product information you should submit a variation application as appropriate.
10.2 Standalone PAM submission route
Information concerning PAMs which is not submitted as part of other procedures and does not impact the product information should be submitted to the MHRA as a post-authorisation commitment via the MHRA portal using the information update route, clearly stating the nature of the PAM.
10.3 Assessment of PAMs
Where PAM applies in the EU we will take into account the EU decision and the submission should be made to us be made once the CHMP opinion is available. The CHMP opinion and other relevant EU documentation (AR assessment report etc. where available etc.) should be provided as part of your submission. However, where new information might influence the evaluation of the benefit and risks of the product you are obliged to inform us of this. This information should be provided to the MHRA as soon as is reasonably practicable and this includes where the information comes to light as part of an EU assessment (please refer to section 11 for further information).
In most circumstances the MHRA will not issue a separate approval for information submitted as a standalone PAM unless we have additional requirements, or the PAM is at the request of the MHRA.
11. Implementation of outcomes of EU safety referrals and procedures concerning PSURs, PASS, signal assessments and PAMs
You are reminded that you must ensure that the information for your product is kept up to date with current scientific knowledge. MAHs should continue to monitor the EMA and Heads of Medicines Agencies (for CMDh outcomes) websites and implement the outcomes from EU decisions as appropriate. Where you hold a UK MA which covers Northern Ireland, the EU outcome should be implemented, unless you are advised otherwise, and the same type of variation procedure as recommended in the EU procedure should be used to implement the change for the UK MA.
For Great Britain-only MAs, the expectation is that you will follow EU outcomes and make the same changes to the Great Britain-only MA as made to the corresponding EU MA. In order to do so you will need to submit the corresponding procedure to the MHRA.
In exceptional circumstances, the MHRA may consider that your MA should not be updated in line with the EU decision, where this applies we will inform MAHs within 14 days of the publication of the EU outcome where this is the case. In all other circumstances, an appropriate variation should be submitted.
Where the outcome for the corresponding centrally authorised product is implemented as part of the EC Decision without a separate variation, unless you are advised otherwise, a Type IAIN variation should be submitted to implement the same outcome for the Great Britain-only MA. Where we have UK specific requirements for submission of procedures these are detailed in the relevant sections of this guidance.
Unless otherwise stated in this guidance (please see specific advice regarding PASS and referrals) the reliance route can be used to implement the outcome for the corresponding EU procedure for a Great Britain-only MA. In this case you must provide all relevant EU documentation (CHMP opinion, assessment report and, where applicable, the EC decision). Reliance route applications should normally be submitted once there is a CHMP opinion, and your application should include the CHMP opinion and the corresponding assessment report. Where an EC decision is required, we will not conclude the application until you have submitted this to us.
However, the reliance route is not a substitute for MAH obligations to submit pharmacovigilance data and information to the MHRA and to keep the MA up to date with current scientific knowledge. Where there is new information which might impact the evaluation of the benefit and risks of your product, is likely to impact clinical management of patients and/or require proactive communications (e.g., via a DHPC), including information in the EU assessment this should be submitted to us as soon as possible, and you should not wait until the CHMP opinion is available (also see section 12 regarding safety communications).
Where the MHRA assessment results in additional UK-specific requirements we will contact MAHs directly before or shortly after (within 14 calendar days) the publication of the EU outcome. However, where the necessary information, as outlined above, has not been provided this may lead to a delay in us contacting you regarding UK specific requests.
12. Safety communications
The principles of safety communication outlined in GVP module XV continue to apply to all UK authorised products. The responsibilities of MAHs in relation to public announcements on pharmacovigilance concerns, in relation to the use of medicinal products authorised in Northern Ireland and products authorised in Great Britain-only are set out in the guidance note on the exceptions and modifications to the EU guidance on good vigilance practices. MHRA would like to be made aware of any planned communications as soon as possible and MAHs should provide us with all available documentation, including that from the EU assessment, where appropriate. We consider this to be part of your obligation to inform us where there is any new information which might impact on the evaluation of benefits and risks.
12.1 Direct healthcare professional communications (DHPCs)
Guidance for marketing authorisation holders on drafting direct healthcare professional communications (DHPCs) can be found at How to draft a DHPC
Irrespective of the route of authorisation, the content of a DHPC and communication plan should be agreed with the MHRA before dissemination in the UK. Draft DHPCs should be sent to RMPallocation@mhra.gov.uk
Where the DHPC is a result of an EU assessment, to avoid delay in the dissemination of the DHPC in the UK, as stated above you should inform us as soon as you are aware that a communication may be required and supply all necessary information when available. Once a core EU DHPC, setting out core EU messages, is agreed the final version of the DHPC and communication plan should be sent to the MHRA for review. You should wait until you receive comments and agreement from the MHRA before disseminating the DHPC in the UK.
The timing of dissemination may be adapted according to the urgency of the situation.
13. Requirements for MAs granted via the Unfettered Access route
Where you have a Great Britain-only MA granted via the unfettered access route, all pharmacovigilance obligations continue to apply. To ensure the safety of patients in Great Britain you must provide any new information that may impact on the terms of the MA and/ or impact on the balance of benefits and risk of the product. This includes information from clinical trials and data on the use of the product outside the terms of the MA. Unless otherwise stated, the same data/submission as that provided to the EU should be submitted to the MHRA. Other requirements are set out below
13.1 Submitting ICSRS
You should submit to the MHRA reports of all serious suspected adverse reactions that occur in the UK and other countries, and all non-serious suspected adverse reactions that occur in the UK via the MHRA Gateway and/or ICSR submissions portal.
13.2 Provision of other Pharmacovigilance data
You should submit copies of the Periodic Safety Update Reports (PSURs) to the MHRA portal. We will accept copies of the PSUR submitted to the EU in line with the frequency set by the EU.
All other pharmacovigilance data submitted to the EU, including RMPs, PASS protocols and final study reports etc. should also be submitted to the MHRA, and we will accept copies of the same information submitted to the EU.
Contact
For further information, please email our Customer Services Centre at info@mhra.gov.uk or call 020 3080 6000. You can also email vigilanceservice@mhra.gov.uk with urgent questions. Alternatively, contact your Trade Association by emailing:
- Association of the British Pharmaceutical Industry (ABPI): regulatory@abpi.org.uk
- British Generic Manufacturers Association (BGMA): info@britishgenerics.co.uk
- BioIndustry Association (BIA): regulatory@bioindustry.org
- Clinical & Contract Research Association (CCRA): mail@ccra.org.uk
- Ethical Medicines Industry Group (EMIG): info@emig.org.uk
- Health Food Manufacturers’ Association (HFMA): pennyviner@btconnect.com
- The National Pharmacy Association (NPA): independentsvoice@npa.co.uk
- Proprietary Association of Great Britain (PAGB): regulatory@pagb.co.uk