Requirements of the manufacturer's PMS system
Updated 5 September 2025

© Crown copyright 2025
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/medical-devices-post-market-surveillance-requirements/requirements-of-the-manufacturers-pms-system
Requirements of the manufacturers PMS system (Regulations 44ZE, 44ZF, 44ZG, 44ZL, 44ZM and 44ZQ)
The new set of regulations The Medical Devices (Post-market Surveillance Requirements) (Amendment) (Great Britain) Regulations 2024 amends the UK Medical Devices Regulations (MDR) 2002 by inserting a new Part 4A on post-market surveillance (PMS) requirements for medical devices, including in vitro diagnostic (IVD) devices and active implantable medical devices which apply within Great Britain (GB). It includes notification requirements for incidents, and preventive and corrective actions taking place after the device is first approved for the GB market.
Regulations 44ZE, 44ZF, 44ZG, 44ZL, 44ZM and 44ZQ require manufacturers to have a process in place for gathering and analysing feedback and complaints, and to ensure their devices continue to meet appropriate standards of safety and performance.
Manufacturers must use PMS data as one of many sources of input to their risk management process, and to update the technical documentation for UKCA marked devices. Similarly, the output from the device technical documentation, including risk management, provides input to develop the PMS plan. The MHRA expects manufacturers to apply the same principles to update technical documentation for CE-marked devices placed on the GB market, although this cannot be mandated under regulation 44ZE(5).
This regulation requires manufacturers to continually gather information on the performance and safety of their device throughout the PMS period. The manufacturer must determine the device lifetime in order to specify the PMS period in their PMS plan.
Manufacturers should not stop undertaking any of the PMS activities laid out in this guidance at the end of the PMS period because new safety or performance issues can continue to arise at any point the device remains in use. Although not mandatory, this is strongly encouraged by MHRA to allow the manufacturer to identify and address risks identified after the validated use period. Knowledge of these issues provides real-world data that can help protect future device users by enabling manufacturers or regulators to provide advice or take actions to reduce these device-related risks. As the MHRA develop the future medical devices regime, we will consider further consultation on the expansion of these requirements commensurate with device risk.
The MHRA recognises that that the manufacturer can only validate the performance and safety of their device for the specified device lifetime. The purpose of extending post-market surveillance is to gain important insight into device performance beyond the known and characterised period, which could be used to enhance patient safety.
The manufacturer must define their PMS system within a PMS plan, and the plan should be proportionate to the risk posed by the device. Information is available in the section on PSUR on the broad range of activities and data sources from which a manufacturer can obtain feedback on their medical devices.
As a minimum the plan must specify the manufacturer’s:
- objectives of the PMS system
- processes to gather information (ensuring comprehensive real-world data obtained)
- methods of data analysis
- methods used to demonstrate fulfilment of their GB vigilance reporting obligations
- links to preventive and corrective action as part of a risk management process
In the case of system or procedure packs, the manufacturer placing these on the market or putting them into service should ensure they focus on gathering and analysing PMS information relating to the safety and performance of the combined use of the devices in the pack. All PMS requirements apply to those manufacturers assembling the packs with the exception of PMSR or PSUR obligations under certain circumstances. See Reporting against the PMS Plan (Regulation 44ZL or 44ZM) for further information.
The manufacturer must review action taken according to the PMS plan at regular intervals. They must document the review within a PMSR or PSUR as appropriate.
The UK responsible person (UKRP) must ensure they immediately inform the manufacturer of any complaints or reports they receive relating to medical device for which they have been appointed.
See Figure 1 for PMS System and notification requirements.
Refer to the latest version of PD CEN ISO/TR 20416:2020 ‘Medical Devices. Post-market surveillance for manufacturers’ for further guidance on the PMS process.
Although the minimum document retention period is 15 years for implantable devices, and 10 years for all other devices, all documentation relating to PMS must be retained to the end of the entire PMS period if this exceeds these times (44ZQ).
Important changes introduced by the PMS regulations
- requirement for a PMS plan is now mandatory in the regulations.
Figure1. Manufacturer’s PMS system
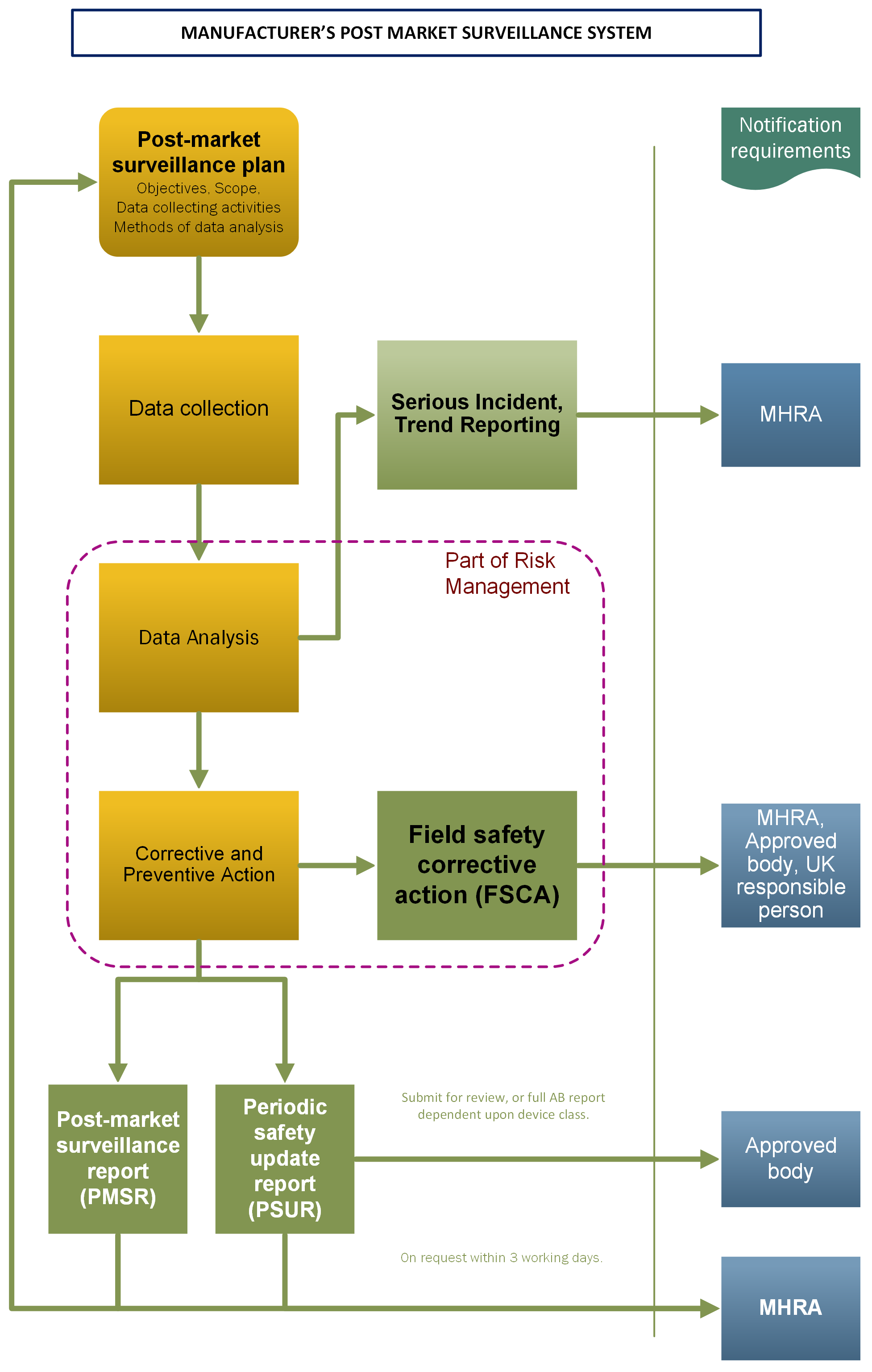
a) PMS plan (Regulation 44ZF)
Full details of what must be covered in the PMS plan are set out in regulation 44ZF of the Regulations. Clarification of certain aspects only is provided below.
i. Feedback from users including patient and public engagement (Regulation 44ZF(3)(a)(v))
Manufacturers must proactively seek feedback from different user groups, including healthcare professionals and patients where relevant. It’s very important to gain information on device safety and performance from patients and the public where appropriate to ensure their views and experiences are captured.
This includes obtaining feedback on usability of the device, and adequacy of the instructions for use which are provided. Manufacturers need to assess and document the extent to which this is relevant and achievable dependent upon the particular device type. The methods used to collect information should be tailored to the populations who typically use the devices. Although not limited to these devices, this is particularly important for devices used by patients or members of the public with limited or no involvement from healthcare professionals. For these devices, direct contact with patients and the public may be required to gather data on product performance and safety.
The manufacturer should consider the most suitable and achievable ways to capture this information and the necessary frequency of this activity, facilitating feedback in the least burdensome way for users, dependent upon the type of device and its circumstances of use (for example, over the counter devices, devices used at home and/or by vulnerable populations). Where there is a reluctance to share information, manufacturers should review why this is, what the barriers for customers are and how they can be addressed.
For further information and advice on undertaking patient and public engagement, see the documents listed below which share ideas on transferable concepts to support these activities:
ii. Safety information on similar devices, including competitors’ devices (Regulation 44ZF(3)(a)(vi))
Timely and effective signal detection relies on manufacturers reviewing global data on device performance and safety. Safety issues arising with one type or model of device may have serious implications for the safety of similar devices.
Similar devices
The regulation explains that similar devices are those based on the same or similar technology and with the same or similar intended purpose.
The manufacturer’s PMS system must therefore include the collection of data on the performance of their device across all markets, and of publicly available information on the safety of competitors’ devices which may have an impact on their own.
Where there are many similar devices on the market, the manufacturer’s device should be compared to devices that not only have the same intended purpose but also the same intended use environment. For example, pregnancy tests are used both within and outside healthcare settings and those for professional use should also be considered relative to their intended purpose and context (for example, emergency department, pre-operative assessments).
Important changes introduced by the PMS regulations - PMS plan
- clarification that feedback should include patient and public engagement
b) Preventive and corrective action (Regulation 44ZG)
One important objective of the PMS process is to ensure that the manufacturer identifies the need for and undertakes timely preventive and corrective action whenever necessary to protect the safety of those affected by the use of their medical devices. The findings from the PMS of one type or model of device may have implications for the safety and performance of other device types or models which share common characteristics.
Manufacturers must perform effective and continuous risk analysis, evaluation and management for their devices, which may be through adherence to the latest version of the Risk Management Standard, ISO 14971: Medical devices — Application of risk management to medical devices. Risk management includes the requirement to identify and reduce safety risks as far as possible, and covers all safety risks, not only those which present a risk of a serious incident.
Preventive actions are taken to eliminate the root causes of potential non-conformities and risks identified during the PMS period, to prevent their occurrence. This includes preventive actions for ongoing manufacturing of a product line where a risk or non-conformity has been identified during post market surveillance. Before completion of the manufacturing process, manufacturers must:
- take preventive action to reduce risks that could arise when the device is used, or
- remove the cause of any non-conformities with the essential requirements
This includes a vast range of measures including, but not limited to, device manufacturing process corrections, improvements, or design changes.
Corrective actions are required if safety or conformity issues arise among devices after they have been manufactured. These actions are taken to eliminate the cause of a non-conformity or identified risk posed by a device to prevent reoccurrence. This applies to devices stored within the manufacturer’s control (for example, in a warehouse) as well as those which have already been distributed outside the manufacturer’s control.
Corrective action to address a risk of a serious incident affecting devices outside the manufacturer’s control (that is, within the field) is known as field safety corrective action (FSCA). The section on FSCA explains that these can include a wide range of actions, in addition to device recall or withdrawal from the market.
If all the following criteria apply, then the corrective action constitutes FSCA:
- field: devices have already been made available for use (for example, entered the distribution chain to a separate distributor, importer, retailer, hospital, or provided directly to the end user
- safety: there is a risk of death, serious deterioration in health or serious public health threat
- corrective action: any action taken to reduce or mitigate this risk for the devices
The UK-based manufacturer or the UKRP must co-operate with the MHRA on any preventive or corrective action taken to reduce risks posed by medical devices.
Manufacturer’s notification requirements
To UK responsible persons (UKRP) and UK approved bodies (UKAB)
Manufacturers must inform their UKRP and/or their UKAB (where they have them) of all preventive and corrective actions taken after device certification for the GB market to address a risk or non-conformity compromising the performance or safety of the device, regardless of how the risk was identified (regulation 44ZG(2)). This enables the UKAB to assess whether there is an impact of the device certification they have issued (regulation 44ZG(3)).
The process for provision of this information should be agreed between the manufacturer and their UKAB (as part of the ongoing UKAB surveillance process), or with their UKRP as applicable. There is flexibility to determine the most effective and practical way to do so, and the PMSR or PSUR may provide a useful tool for this purpose.
The MHRA recommends that the manufacturer informs their UKRP immediately if their UKAB cancels or suspends their device certification.
Where a manufacturer undertakes FSCA they should ensure both the UKAB and UKRP are aware in advance of initiating the action (44ZJ)
To the MHRA
Manufacturers must inform the MHRA of FSCA before they are undertaken as part of their corrective action (see section D).
Important changes introduced by the PMS regulations - Preventive and corrective action:
- requirement to inform the UKRP and UKAB (where they have them) of all preventive and corrective actions taken to address a risk compromising the performance or safety of the device, or to address a non-conformity with the relevant essential requirements
- UKAB to review preventive and corrective actions for impact on certification
c) Reporting against the PMS plan (Regulation 44ZL or 44ZM)
The manufacturer must produce either a post-market surveillance report (PMSR) or periodic safety update report (PSUR) for any devices it places on the market or puts into service in GB. These requirements are applied in a manner that corresponds to the risk classification of the device according to the applicable legislation, with higher risk class devices being subject to greater regulatory oversight.
The manufacturer must produce a PMSR (regulation 44ZL) for:
- devices falling under Class I
- IVDs falling under class A or B, or IVDs not on list A or B of Annex II of Directive 98/79
The manufacturer must produce a PSUR (regulation 44ZM) for:
- medical devices falling under Class II or III or
- IVDs falling under class C or D, or on list A or B of Annex II
These reports are not required for:
- custom-made devices (regulation 44ZD(2))
- system or procedure packs where the final device is not UKCA or CE-marked when placed on the GB market or made available for use (Regulations 44ZL(4)(a) and 44ZM(4)(a))
The requirement to produce these reports applies from the date of certification for the GB market, or date of declaration of conformity, throughout the PMS period, even in circumstances where the device certification has expired, or the device has been discontinued. The PMS requirements apply to devices put on the market or put into service from 16 June 2025 onwards. There is no backdating of requirements, therefore the requirement to produce these reports will apply from 16 June 2025 onwards.
These reports must contain data relating to devices placed on the market or put into service from 16 June 2025 onwards. Where devices were on the market prior to 16 June 2025, manufacturers should include analysis of historical data collected through previous post-market surveillance activities as they were conducted prior to the new requirements.
The manufacturer must prepare a PMSR/PSUR regardless of whether they have a UK Approved Body appointed.
The report should provide a comprehensive and critical analysis of the risk-benefit balance of the device, in the context of cumulative information gathered since the report was first written. It should be a standalone and searchable document in the technical documentation on PMS drawn up by the manufacturer, but which can be assessed independently.
As a minimum, these reports must include a summary of the analysis and conclusions from review of the PMS data gathered since the last report, in accordance with the PMS plan. Manufacturers must provide details of any preventive or corrective action taken since the last report, with potential impact on devices safety, performance and quality, including any FSCA, and why such action was taken. If the first individual device was placed on the market or put into service after the 16 June 2025, the first report should include any action taken since device certification or the declaration of conformity.
The main objective of this review is to enable identification of any changes to the benefit-risk profile of the medical devices, considering new or emerging information, enabling transparency on post-market data for MHRA. If the manufacturer identifies concerns, this information should be used to re-evaluate whether the device continues to meet the state of the art for the medical device(s) and what risk reduction action could be taken. Hence the review allows for re-evaluation at defined time-points after market approval as to whether the medical device remains safe and effective.
If the manufacturer is submitting PSURs produced in line with the EU MDCG guidance to an EU Notified Body, MHRA consider this existing PSUR may be updated with information specific to the GB market when submitting to MHRA and the UKAB. The MHRA has published detailed PSUR guidance which outlines what additional detail should be included within the report for GB.
System and procedure packs
The PMSR/PSUR requirements apply to system or procedure packs which:
- contain a medical device which does not bear a UKCA or CE-mark, or
- where the chosen combination of medical devices is not compatible with their approved intended use.
These system or procedure packs shall be classified in line with accepted classification guidance, and the PMSR/PSUR requirements apply accordingly. Specifically, this should be determined by the intended use of the pack, but if in doubt it should align with the highest classified device in the pack where applicable, taking into account the intended use of the final device.
The PMSR or PSUR requirements for these systems and procedure packs apply to the devices when used together in the combination as supplied.
Any device which bears a UKCA or CE-mark and which is supplied as part of a system or procedure pack and separately in its own right requires is own PMSR or PSUR as the data under analysis will not be the same.
Notification requirements to the MHRA
The manufacturer must make the most up-to-date report available to the MHRA on request within 3 working days. There is no requirement to routinely submit copies of PMSRs or PSURs to the MHRA unless requested to do so.
PMSR and PSUR preparation and review requirements
The following tables summarise the requirements for preparation of the PMSR and PSUR.
Table 1. PMSR preparation and review requirements
| Device Classification | Special subgroups | Involvement of UKAB in PMSR /PSUR | Method of Provision to MHRA | Minimum frequency of update |
|---|---|---|---|---|
| Class 1 | General (exclude those in Class I below) | None required | On request | Every 3 years |
| General IVD (not Annex II list A&B) | General | None required | On request | Every 3 years |
| IVD Class A | General (exclude those in IVD Class A below) | None required | On request | Every 3 years |
| Class I & IVD Class A | Sterile, measuring function, reusable surgical instrument | None required | On request | Every 3 years |
| IVD Class B | Not applicable | None required | On request | Every 3 years |
Table 2. PSUR preparation and review requirements
| Device Classification | Special subgroups | Involvement of UKAB in PMSR /PSUR | Method of Provision to MHRA | Minimum frequency of update |
|---|---|---|---|---|
| Class IIa | Non-implantable | Review | On request | Every 2 years |
| Class IIa | Implants (Oral or nasal cavity, ear canal) | Review and complete report | On request | Every 2 years |
| Class IIb | Non- implantable | Review | On request | Every (1) year |
| Class IIb | Implants | Review and complete report | On request | Every (1) year |
| Class III | N/A | Review and complete report | On request | Every (1) year |
| IVD Annex II (List A&B) | N/A | Review and complete report | On request | Every (1) year |
| IVD Class C | N/A | N/A no UKAB (NB review with/without report). May change when future GB medical device regulations are introduced. | On request | Every (1) year |
| IVD Class D | N/A | N/A no UKAB (NB review with/without report). May change when future GB medical device regulations are introduced. | On request | Every (1) year |
d) The post-market surveillance report (PMSR)
The manufacturer must prepare a PMSR within 3 years of the first device being placed on the market or put into service (whichever is sooner), or if already on the market/put into service, within 3 years of this regulation coming into force.
The manufacturer should update the report when required but at least every 3 years. This provides flexibility for the manufacturer to align updates to the PMSR with similar requirements within other regulatory jurisdictions within this 3-year period or to spread the requirement across the year as needed.
Providing the above requirements are met, the format and presentation of the PMSR may be determined by the manufacturer. The presentation and methodology used, however, should be consistent from one PMSR to the next, to enable comparison across reporting periods.
As an overview the PMSR should include:
- a summary of the results and conclusions of the benefit-risk determination based upon review of the risk analysis and evaluation
- a description of any preventive or corrective action taken to address a risk or non-conformity compromising the performance or safety of the device since device certification/declaration of conformity, including FSCA, and the rationale for doing so
The MHRA has published detailed PMSR guidance which provides some useful suggestions on how to present information which manufacturers may find helpful in preparing the PMSR.”
e) The periodic safety update report (PSUR)
The PSUR should not duplicate all data and reports generated by the PMS plan, but provide an overview of the PMS activities, data collection, analysis results and summary of conclusions.
The manufacturer should prepare the PSUR according to a standardised format to enable a consistent method for reporting from one manufacturer to another for these higher-risk devices. Further advice is available on the format and presentation of data which should be followed as far as possible for all device classes. If a manufacturer decides that specific sections or datasets are not required, they should document the justification in the PSUR.
The MHRA has published further guidance on the format and presentation of data in our PSUR guidance.
As an overview the PSUR should include:
- a summary of the results and conclusions of the benefit-risk determination based upon review of the risk analysis and evaluation
- the supporting risk analysis and evaluation
- the findings and conclusions of post-market clinical follow-up (PMCF)
- a description of any preventive or corrective action taken to address a risk or non-conformity compromising the performance or safety of the device since device certification/declaration of conformity, including FSCA, and the rationale for doing so
- the volume of sales/supply in the UK
- an estimate of the size of the population using the device inside and outside the UK and the usage frequency, where practical
- the characteristics of the population using the device including details of any higher-risk sub-populations
To prepare the above summary, the manufacturer should consider the following elements:
- information concerning serious incidents, including those arising from side effects having a negative impact on the health of the patient, their care or on wider public health
- information from trend reporting
- information relating to non-serious incidents
- relevant specialist or technical literature, databases and/or registries
- information, including feedback and complaints, provided by users including patients and the public, distributors and importers
- publicly available information about similar medical devices inside or outside GB
The types of data sources available may differ. Manufactures should adapt and justify within their PMS plan and PSUR.
Timescales for provision of PSURs by device risk classifications
See PMSR and PSUR preparation and review requirements above.
Class IIa
The manufacturer must produce the first PSUR within 2 years of the device being placed on the market/put into service or if already on the market within 2 years of this regulation coming into force (see saving provision referenced above).
Update the PSUR every 2 years throughout the PMS period.
Class IIb and Class III medical devices and Class C and D, and Annex II (list A and B) IVDs
The manufacturer must produce the first PSUR within 1 year of the device being placed on the market/put into service or if already on the market within 1 year of this regulation coming into force (see Saving Provision referenced above).
Update the PSUR every year throughout the PMS period.
Involvement of the approved body (UKAB)
See PMSR and PSUR preparation and review requirements above.
The manufacturer must submit their PSUR (and updated PSUR) to their UKAB if they have one, according to pre-agreed arrangements.
For class C or D IVDs, for which there would be no UKAB, the EU requirements to submit the PSUR to the notified body should be followed. This aspect cannot be covered by this GB regulation please refer to MDCG guidance.
The UKAB must take the PSUR into account as part of its assessment of the ongoing validity of certification they have issued for the device(s).
The PSUR should form part of the documentation which the UKAB reviews within surveillance activities relating to conformity assessment. This may include a sampling-based approach, depending on the device classification and assessment route chosen for the device.
For class III, all implantable devices and Annex II list A and B IVDs, the UKAB must prepare a report on its conclusions from its review of the PSUR. The report should determine whether there is any impact on the validity of the device certification it has issued, and if so, provide details of any action that is needed. The time required to complete this report may vary depending upon the complexity of the PSUR, but it should be finalised as soon as reasonably practical. The MHRA has published further guidance on the UKAB report in our PSUR guidance.
Notified bodies should follow EU requirements for review of PSURs, as this aspect cannot be covered by this regulation.
Grouping of devices
For PSUR guidance, the term ‘device’ relates to a device model and not to an individual device, as individual devices are placed on the market at different moments during the period covered by the device certificate. A device should be associated with one basic UDI-DI when UDI is used (or one device where UDI is not available) and may include different variants or sizes.
The manufacturer may prepare a single PSUR for a category/group of devices if they are covered by a single clinical evaluation or performance evaluation report and/or are considered similar devices. Where devices are grouped together, the manufacturer should justify the combination, and the data should be presented in a clear, organised manner so that it is easy to determine how each device and/or device variant performs independently.
The grouping of devices in one PSUR is only possible for devices for which the conformity assessment activities have been conducted by the same UKAB. This is to facilitate the review and evaluation process by the UKAB. UK CA-marked and CE-marked devices may be grouped together providing other grouping requirements are met, and they are within the remit of the same conformity assessment body.
For devices, including the leading device, which have been on the market with subsequent certificates of different UKABs, the revision history provided should make a reference to the previous PSUR versions where the former UKAB(s) were involved and, when applicable, indicates the actions they required or undertook.
The ‘leading’ device
For devices grouped together in the same PSUR, the manufacturer should assign a ‘leading device’ which needs to be the highest risk class or one of the highest risk classes. The PSUR reference number is attached to the leading device and should remain unchanged for the PSUR updates, provided the leading device in the grouped devices has remained the same.
The leading device determines the schedule for the data collection period and PSUR update/ frequency applicable to the whole group of devices irrespective of the device class or certification date for the other devices.
When a device grouping has been established, it could be amended for the PSUR update(s) by removing or adding devices except for the leading device, which cannot be changed.
The manufacturer should provide justification for the change, together with the PSUR reference number of the PSUR where the data of the removed device(s) are reported.
In case of a change related to the leading device (new device model /change of the basic UDI DI), a new PSUR should then be issued.
PSUR updates for the group of devices which includes the former leading device should continue independently for the PMS period of the former leading device.
Important changes introduced by the PMS regulations - Reporting against the PMS plan:
- provision of PMSR or PSUR to MHRA on request within 3 UK working days
- deadline for preparation of first PMSR and update at least every 3 years
- preparation of a PSUR to a standardised format, updated at set intervals
- submission of PSUR to approved body
- UKAB to review PSUR for any impact on device certification
- UKAB to prepare a report on review of PSUR for certain device classifications
f) Reporting under the GB Medical Devices Vigilance System (Regulations 44ZC, 44ZH, 44ZI, 44ZJ, 44ZK, 44ZN, 44ZO and 44ZP)
The Medicines and Healthcare products Regulatory Agency (MHRA) is the regulatory authority for the UK medical device market. Once a medical device has been placed on the UK market, the manufacturer must submit reports to the MHRA when incidents that involve their device occur in the UK and meet specific criteria.
The manufacturer must also take appropriate action to address safety risks when required. This includes addressing risk relating to devices which they have already sold or made available for use. These actions are known as field safety corrective actions (FSCAs).
The notification and evaluation of serious incidents, trends and FSCAs involving medical devices is known as the GB medical device vigilance system.
MHRA has provided guidance on how and what to report under the GB vigilance system.