Guideline IV Signal management, including benefit-risk reports
Updated 29 May 2024
Applies to England, Scotland and Wales

© Crown copyright 2024
This publication is licensed under the terms of the Open Government Licence v3.0 except where otherwise stated. To view this licence, visit nationalarchives.gov.uk/doc/open-government-licence/version/3 or write to the Information Policy Team, The National Archives, Kew, London TW9 4DU, or email: psi@nationalarchives.gov.uk.
Where we have identified any third party copyright information you will need to obtain permission from the copyright holders concerned.
This publication is available at https://www.gov.uk/government/publications/pharmacovigilance-of-veterinary-medicines-in-great-britain/periodic-safety-update-reports
1. Signal management process
1.1 Signal management process overview
Signal management is performed to detect potential safety signals and investigate the possibility of a previously unidentified potential risk, a change in the status of a known risk, or provide reassurance about the absence of a risk of a product or active substance.
A veterinary adverse event signal is information that arises from one or more sources which may suggest a new potentially causal association, or a new aspect of a known association, between an adverse event or set of related events and one or more veterinary medicinal products (VMPs) or active substances. The signal may involve a previously unknown event or could involve an event reported with a higher frequency than what is expected in a population. It may warrant further investigation and, when necessary, action. A signal does not always mean that a VMP has caused the suspected adverse event. Assessment of the signal is required to determine whether or not there is a causal relationship.
Signals may also be identified in relation to lack of efficacy or development of resistance.
Signals may originate from sources such as spontaneous reports, clinical, non-clinical or epidemiological studies, and published literature. They may be identified where:
-
a sudden increase in the number of adverse events in a short period is observed
-
an increase in the frequency of a particular clinical sign or Veterinary Dictionary for Drug Related Affairs (VeDDRA) Preferred Term (PT) is recorded, compared with the expected frequency for that sign
-
an increasing trend of a particular clinical sign or VeDDRA Preferred Term is noted over time
-
new, previously unidentified, clinical signs or VeDDRA PTs are identified
-
a potential impact on, or risk to public or animal health, or welfare is suspected
New information that may be relevant to the signal management process may include the severity, time to onset, duration or outcome of an adverse event, the country in which the event occurred, or clinical information such as gender, age, breed, cause of death or off-label product use.
Signals can relate to an active substance, a particular VMP, or a group of related active substances or VMPs. They may also relate to a specific strength or formulation of a VMP.
Multiple spontaneous adverse event reports are usually required for the threshold to be met for a signal. If a single report contains detailed information regarding an adverse event of significant seriousness or severity and/or involved multiple animals, it may be considered to meet the threshold, but this would only be applicable in very rare and specific circumstances where there is considered to be a significant risk to human, animal, or environmental health or welfare.
1.2 Signal management process requirements
Both the VMD and MAHs are responsible for detecting and managing signals.
The obligations of the Marketing Authorisation Holder (MAH) for signals associated with their VMPs for which Marketing Authorisations (MAs) are held are defined in the VMR 2013 (as amended) Schedule 1 Part 8 paragraph 56 and 59.
The VMR 2013 (as amended) defines the terms benefit-risk balance and signal management process which are referenced in this guideline and can also be found in the Glossary.
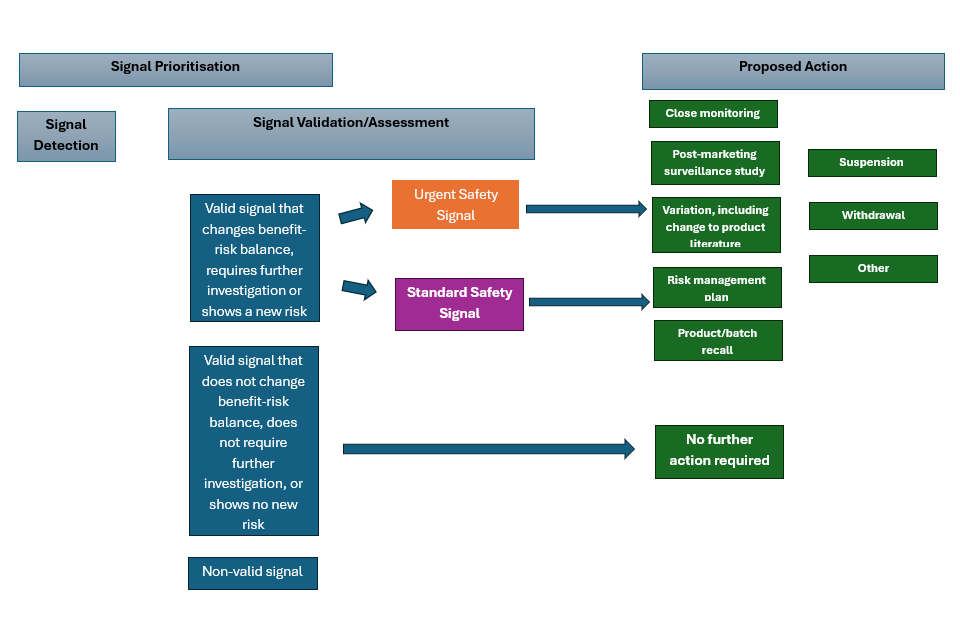
The signal management process is a process for performing active surveillance of pharmacovigilance data for VMPs and active substances, assessing that data, and determining whether there is any change to the benefit-risk balance of those products or active substances.
The process should include signal detection and analysis, validation, prioritisation, assessment and confirmation, and subsequent recommendation of proposals for action if required. The order in which these steps are taken may depend on the data available to the MAH. A risk-based approach should be utilised.
The benefit-risk balance is an evaluation of the positive effects of a VMP in relation to risks to human or animal health (relating to the quality, safety, or efficacy of the product), risks of undesirable effects on the environment, or any risk relating to the development of resistance.
MAHs should continuously monitor the benefit-risk balance of their VMPs via a documented signal management process, whether the reports derive from the UK or any other country. This allows for any emerging issues that may affect the benefit-risk balance to be promptly detected.
Both documentation detailing internal signal management processes and evidence that these processes have been followed should be promptly provided to the VMD upon request, whether in relation to an inspection or at any other time.
At a minimum the process should be able to identify:
-
a sudden and unexpected increase in the number of adverse events
-
an unexpected increase in the frequency of a known clinical sign
-
a new clinical sign
-
reports in scientific literature of any of the above
The MAH must record and submit the results of this signal management process on an annual basis to the VMD via a Benefit-risk report (BRR) and Signal notifications (see sections 2, 3 and 4 of this guideline).
If a validated signal suggesting a new risk or change of the benefit-risk balance of the product, or where further investigation/monitoring is required, is identified during the process, the MAH must notify the VMD promptly and within 30 calendar days of it being identified. If an urgent safety signal is identified, the MAH should notify the VMD without delay and no later than the next working day for signals that require restrictions to be implemented or 3 days where no restrictions are deemed necessary (see section 2). Where the process identifies the necessity for a variation in an authorisation, the MAH must also promptly submit an application for such a variation.
Any additional validated signals that do not suggest a new risk, change to the benefit-risk balance or that do not require further investigation or monitoring following assessment, should be submitted as part of the BRR.
The MAH is obliged to inform the VMD of any valid signal identified by any regulatory authority in any country where the product is marketed which might influence the evaluation of the benefit-risk balance of the product concerned.
Notification of a validated signal, urgent safety signal, or signal identified by another regulatory authority should be carried out via completion of the signal and regulatory actions section of the BRR/Signal template (see section 3 and 4.7 of this guideline).
1.3 Signal detection and analysis
MAHs should aim to utilise all relevant post-marketing pharmacovigilance data which they could reasonably be expected to be aware of, including:
-
data collected within a pharmacovigilance database, such as:
- spontaneous reports obtained via direct reporting to the MAH from all reporters, including those received via sales representatives or other company employees,
- spontaneous reports obtained as part of an enquiry or product defect concern,
- spontaneous reports that the MAH is aware of reported on social media or other media sources,
- spontaneous and solicited reports received from other MAHs, manufacturers, or regulatory authorities,
- reports from routine searches of published literature.
-
post-marketing trials, post-marketing surveillance studies or other observational studies
-
new toxicology and international safety data updates
-
sales data
Increasing the number and variability of data sources, and additional focused data analysis of certain product groups or patient demographics may additionally benefit the signal detection process.
Signals may be detected via qualitative review of individual spontaneous adverse event reports or using semi-quantitative and quantitative statistical analysis methods. A combination of both methods is generally preferable, although the method(s) used may depend on the volume of reports received by, and the size of, the MAH.
When a large number of adverse event reports are received or multiple data sources are integrated, it is recommended to use statistical methods and data mining algorithms based on 2X2 contingency tables producing disproportionality analysis.
Essentially, this provides a measure of the risk (for a particular product/active substance/product group) of an event being reported versus other events, compared to a reference risk (that observed for all products/active substances in the database or for all other products in that product group).
Frequently used measures of association include proportional reporting ratio (PRR), reporting odds ratio (ROR), relative risk reduction (RRR), and information coefficient (IC). The individual approach of an MAH is considered best selected depending on the database available.
1.4 Signal prioritisation
Signals should be prioritised according to their likelihood of having a significant impact on individual, population, environmental, or public health. Prioritisation should take into consideration the benefit-risk balance of a product or its active substance and those most likely to lead to risk minimisation measures or regulatory actions, particularly those that might need to occur in a timely fashion.
All events that occur in humans and all events related to lack of efficacy should be prioritised.
The VMD has included in section 1.4.1 a list of Medically Important Terms (MITs) which includes VeDDRA PTs that are considered significant medical concepts and should be used for signal prioritisation. This is a non-exhaustive list and subject to update. Some MITs are species-specific.
Prioritisation should additionally consider the frequency of the event, severity of the event, population risk (determined by the size of the exposed population), type of medicinal product/active substance, the length of time the product has been on the market, and the amount of known pharmacovigilance data over the lifecycle of the product.
In certain situations, signals that may be linked to topical public concerns or media attention may need to be prioritised, if there may be a benefit to the release of prompt communications.
Signal prioritisation should be carried out throughout the signal management process.
1.4.1 List of Medically Important Terms (MITs)
This section provides a list of MITs at the level of VeDDRA PT. This list will be regularly updated and therefore MAHs should always ensure that the latest version is being used. It is intended to be used as guidance for prioritisation and analysis of data during the signal management process, however, absence of an event from this list does not exclude that event from analysis.
| PTs | Species |
|---|---|
| All | Human |
| Abdominal pain | Horse |
| Abomasitis | Ruminant, Camelid |
| Abortion | All |
| Acute mastitis | Ruminant, Camelid, Horse |
| Aggression | All |
| Anaphylaxis | All |
| Anorexia | Horse |
| Apnoea | All |
| Ataxia | Horse |
| Bee systemic disorder NOS | Bee |
| Birth defect | All |
| Blindness | All |
| Bone marrow hypoplasia | All |
| Cardiac arrest | All |
| Cardiac insufficiency | All |
| Circulatory shock | All |
| Coagulopathy | All |
| Collapse NOS | All |
| Coma | All |
| Convulsion | All |
| Deafness | All |
| Death | All |
| Diabetes mellitus | All |
| Disseminated intravascular coagulation | All |
| Dyspnoea | All |
| Epileptic seizure | All |
| Fish asphyxia | Fish |
| Fish body deformity | Fish |
| Haemolytic anaemia | All |
| Haemorrhagic gastroenteritis | All |
| Heart block | All |
| Hepatic failure | All |
| Hypersensitivity reaction | All |
| Hypocalcaemic condition | Ruminant, Camelid |
| Hypomagnesaemic condition | Ruminant, Camelid |
| Impaired hearing | All |
| Impaired vision | All |
| Immune mediated thrombocytopenia | All |
| Increased coagulation time | All |
| Ketosis | Ruminant, Camelid |
| Laminitis | Horse |
| Loss of consciousness | All |
| Lying down | Horse, Ruminant, Pig, Camelid |
| Metastatic neoplasia | All |
| Metritis | All |
| Moribund | All |
| Multi-organ failure NOS | All |
| Myoglobinuria | Horse |
| Paralysis | All |
| Paresis | All |
| Perinatal mortality | All |
| Recumbency | Horse, Ruminant, Pig, Camelid |
| Renal insufficiency | All |
| Reticulitis | Ruminant, Camelid |
| Stillbirth | All |
| Suspected infectious agent transmission | All |
| Thrombocytopenia | All |
1.5 Signal validation
An initial validation step should be carried out to determine whether more detailed analysis is justified.
As far as possible, it should be ensured that the signal is not based on duplicate reports.
There should always be a temporal association (the event must have occurred after exposure to the product occurred).
Signals related to adverse event terms (or clinical signs that can be considered covered by the wording) already present in the product information, should generally be counted as non-valid signals. However, if a signal highlights an additional aspect of a current association that may warrant alteration of the wording (for instance regarding the reversibility or expected duration of an event) or to the frequency category, it may be a valid signal.
Signals involving MITs (see section 1.4.1) that are already present in the product information should also be considered valid, however may only warrant a limited assessment (such as calculation and comparison of incidence).
1.6 Signal assessment and confirmation
Once a signal has been validated, further evaluation should be carried out to determine whether or not there is a possible causal association, or new aspect of a known association between the product and event, and whether the evidence is strong enough to consider further action.
Assessment should ideally involve review of all data available, for example from spontaneous reports, literature reports, pre-clinical trials and clinical studies, toxicology and epidemiological updates, international regulatory data, and sales data. All previously reported cases including the same events and products/active substances should be reviewed as applicable. In some cases, it is recommended to additionally review any previously reported cases involving similar events and similar products/active substances.
The evaluation should provide clinical context to the report and take into consideration other knowledge of an association (previous or concurrent reports, previous signals, previous analysis via Periodic Safety Update Reports (PSURs)/BRRs for example).
How strong the evidence is may also depend on the total number of reports, the source of the reports (such as if they are from one or different sources, the quality of the source, and if the data is incomplete or vague), and the availability of supportive laboratory data. The potential for over-reporting should also be taken into consideration, for example due to increased media attention, or a known significant increase in sales volumes.
The following list contains considerations that may be made when assessing signals:
-
total number of cases (excluding duplicates)
-
increase in the number of reports (incidence)
-
clinical relevance (seriousness, severity, outcome, reversibility, relationship to signalment)
-
dose-adverse event relationship
-
drug-drug interaction
-
the consistency between reports (for instance in terms of time to onset, pattern of events, outcome, dosages involved)
-
plausibility of the pharmacological/biological mechanism linking the product and event, or a lack of alternative causes
-
dechallenge/rechallenge data
-
supportive relevant investigation/laboratory data
-
clinically similar events occurring in additional reports
-
other potentially impacting clinical variables such as concurrently administered products, medical history
-
potential for over-reporting, for example due to increased media attention, or a known increase in sales volumes
Following assessment of a signal, a conclusion should be made as to whether or not there is good enough evidence to suggest a potential causal association between the product/active substance and event, in order to determine whether action needs to be taken.
1.7 Recommendations for action
If a potential causal relationship between a product/active substance and event is considered unlikely or there is not strong enough evidence at that time, the benefit-risk balance is considered to remain the same and no action is required at that time. A record of assessed signals that have been determined not to meet the threshold for further action should be maintained for review during subsequent signal assessment and potential re-assessment upon obtaining further information.
If a potential causal relationship is considered possible, it should be determined whether further information is required to provide additional evidence, or whether the benefit-risk balance can be considered to have been altered.
If further information is required, one of the following recommendations for action should be proposed by the MAH, with an explanation of the reasoning behind the proposal:
-
close monitoring
-
post-marketing surveillance study
Close monitoring requires in-depth assessment of all new or updated adverse event reports pertaining to the signal. The VMD may also request additional information to be provided at any time. If this occurs then the reason for the requirement and the time by which, or the period during which, the requirement must be complied with will be discussed with the MAH.
If the benefit-risk balance is considered altered; the MAH should propose risk minimisation measures or other relevant actions as appropriate.
The benefit-risk balance may be improved either by increasing the benefits, for example including further explanation of how best to use the product, or by reducing the risks by risk minimisation measures, for example by contraindicating the use in animals particularly at risk, reducing dosage, or introducing precautions for use. When proposing measures to improve the benefit-risk balance of a VMP, the feasibility of those measures under normal conditions of use should be taken into account. If dose reduction is considered as a method of risk minimisation, the impact of dose reduction on efficacy should be carefully evaluated.
The following types of management actions may be necessary and may be initiated by the MAH or by the VMD:
-
variation of marketing authorisations (MAs) in respect of the indication, dosing recommendations, contraindications, warnings and precautions for use or information about adverse events or other sections of the product literature
-
direct provision of important safety information to veterinarians and other health-care professionals and animal owners, for example through letters, bulletins, via electronic media etc
-
urgent safety restrictions may be taken by MAHs in the event of risk to human or animal health or to the environment. If the MAH implements an urgent safety restriction, the MAH shall give the VMD prior or simultaneous notification. Urgent safety restrictions may also be initiated by the VMD
-
suspension or withdrawal of the MA of a VMP, in the event that, the overall benefit-risk balance is considered unfavourable and proposed risk minimisation measures are considered inadequate. Relevant stakeholders should be informed as appropriate
Such actions may be taken voluntarily by MAHs. However, it is recommended that any such intended measure be discussed at an early stage with the VMD.
If the resulting action requires a variation to an MA, the MAH must promptly submit an application for such a variation, alongside data to support the proposed variation. The VMD will assess, and accept or reject the variation, based on the benefit-risk balance.
The MAH should also inform the VMD of any prohibition or restriction imposed by the regulatory authority of any country in which the VMP is authorised within 30 calendar days of the receipt of such information, if no equivalent measure has been already taken in GB.
2. Urgent safety signals
Urgent safety signals are those containing new information affecting the benefit-risk balance of a VMP which require rapid implementation of risk minimisation/safety measures.
These signals may be identified from studies, spontaneous reports, published literature, or be related to regulatory actions proposed by other regulatory authorities.
Urgent safety restrictions may need to be taken in the event of a risk to human or animal health, or to the environment, and may take the form of an interim change to the product literature or distribution category, for example, therapeutic indications, dosage information, contraindications or warnings, a risk management plan, batch recall or suspension/withdrawal of a product. It may be necessary to provide safety information communications to relevant stakeholders.
If urgent safety restrictions are proposed by an MAH, the VMD must be informed no later than the next working day from when evidence comes to the attention of the MAH of the reasons for the action. The VMD should also be notified of any urgent safety restrictions imposed by other regulatory authorities immediately.
The VMD should otherwise be informed of a potential urgent safety signal not deemed to require restrictions following the conclusion of an internal signal management process within 3 days.
The initial notification of an urgent safety signal leading to an urgent safety restriction should include, within the ‘Evaluation and summary of findings’ field, brief clinical details and an initial assessment of the urgency and potential impact of the signal. It is appreciated that a full assessment cannot be performed within 1 working day, and that further investigation by the MAH/VMD may ultimately conclude that the initial assessment of the signal/impact means that no further action is required.
Any emerging information following this initial notification should be promptly provided to the VMD.
Following further assessment, for both urgent signals requiring restrictions and those deemed no to require restrictions, the MAH should have provided a detailed description of the signal, including information on the source, incidence, and clinical details, alongside an evaluation of the urgency and potential impact. Any proposed actions, or those that have already been taken should also have been clearly detailed.
The VMD will assess the information as provided and discuss the implementation of actions with the MAH.
Urgent safety measures including any related safety communications can be proposed and implemented by the MAH, however the VMD should be informed of these with prior or simultaneous notice. Urgent safety measures may also be proposed by the VMD at any time where there is a significant concern that the benefit-risk balance has altered.
3. Notifying VMD of a signal
3.1.1 Validated signals suggesting a change to the benefit-risk balance
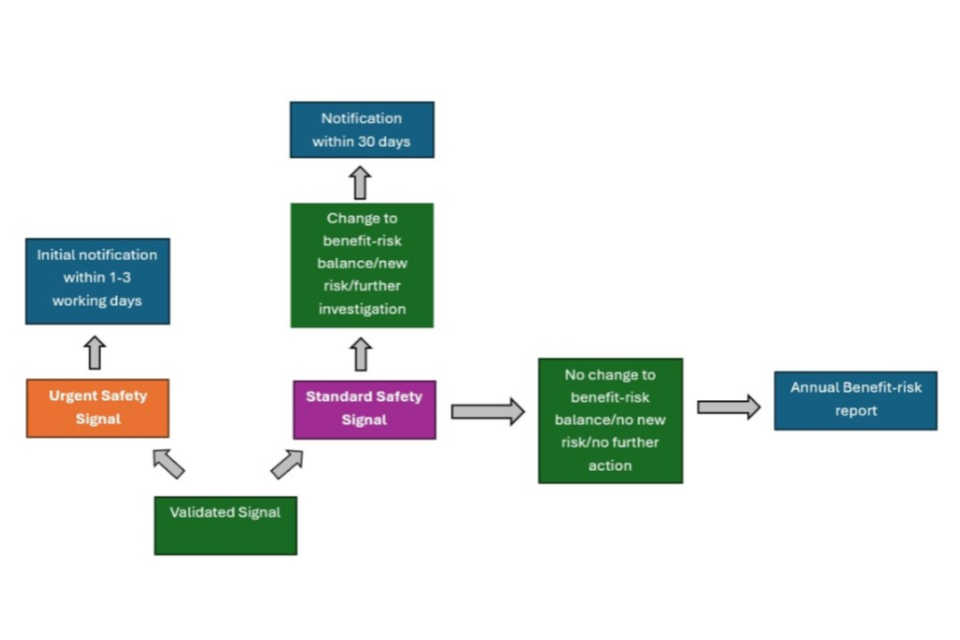
The VMD should be notified without delay of all validated signals which following assessment, suggest a new risk, change to the benefit-risk balance, or require further investigation.
The VMD should be notified of these signals using the Signal notification option or Urgent signal notification option on the BRR/Signal template within 30 calendar days of conclusion of an internal signal management process for standard signals, or no later than the next working day for urgent signals that require restrictions to be implemented or 3 days where no restrictions are deemed necessary. Valid signals identified by any regulatory authority in any country where the product is marketed which might influence the evaluation of the benefit-risk balance of the product concerned should also be reported in this way.
Signals should be accompanied by a proposal by the MAH to either obtain further information (via close monitoring or a post-marketing surveillance study), perform a regulatory action (change to product literature via variation, risk management plan (RMP), product recall, suspension of product, withdrawal of product) or for there to be no further action taken at that time.
A separate signal notification document must be provided for each signal. The signal will be linked to the MA number or Product Group Code stated on the template (see section 3.1.3 and 3.1.4 of this guideline).
A flowchart showing an overview of signal submissions can be found in section 3.1.2 of this guideline.
3.1.2 Validated signals that do not suggest a change to the benefit-risk balance
All validated signals, which, following assessment, are deemed to not suggest a new risk or change to the benefit-risk balance, or that do not require further investigation, should be submitted annually via the BRR template on the ‘Signals and regulatory actions’ tab. Applicable signals that have been assessed via other regulatory authorities should also be included within this section. Signals that have previously been submitted as Signal notifications (urgent or standard) do not need to be re-submitted.
Non-validated signals should be recorded by the MAH but should not be submitted to the VMD.
Multiple signals (adverse event terms) for the same MA number/Product Group Code can be reported within the same annual BRR, but Signal notifications must only contain one signal per notification.
Submission of signals overview
3.1.3 Submission of Signal notifications
Signal notifications should be submitted using the BRR template. Further details on this template and its contents can be found in section 4 of this guideline.
Multiple BRR template documents can be submitted per VMDS submission as long as only one template is submitted per MA or Product Group Code (PGC) (further details on Product Group Codes can be found in section 4.3.3 of this guideline). If further information is requested, or the MAH wishes to submit further documentation, then this can be added to the submission as a separate document.
Signal documents should be submitted should be submitted using the Veterinary Medicines Digital Service (VMDS), a secure messaging service. Any MAH not signed up to VMDS can register at Veterinary Medicines Digital Service.
Once signed in to the VMDS account, the MAH should select the relevant group. Non-urgent ‘standard’ signal notification submissions should be sent to PHV Signals, and urgent safety signals should be sent to PHV Urgent Safety Signals.
The Excel document should be named using the applicable MA number (with an underscore rather than forward slash separation) or Product Group Code (PGC), the year of submission, type of signal (Signal for ‘standard’ signals and Urgent for urgent signals) and number describing the order of signals submitted, separated by underscores: MAnumber_YYYY_Signal_x or MAnumber_YYYY_Urgent_x, or PGC YYYY Signal x or PGC YYY Urgent x.
Number 1 should be used for the first signal for that product of that type submitted in that year, and 2, 3, 4 etc used for subsequent signals. For example, the first standard signal document for a product with an MA number 09285/8019 being submitted in the year 2024, would be named 09285_8019_2024_Signal_1. The second would be 09285_8019_2024_Signal_2. And if subsequently the first urgent signal was submitted for the same product within the same year, the document would be named 09285_8019_2024_Urgent_1.
It is not anticipated that additional documents other than the Excel signal document will be submitted. If further documentation is considered necessary, use the same naming convention as described above, following by underscore 2, 3, 4 for subsequent additional documents. For example, a second document submitted for the above-described urgent signal should be named 09285_8019_2024_Urgent_1_2.
Queries related to urgent safety signal submission or requests for assistance with VMDS in relation to urgent safety signals should be sent by e-mail to adverse.events@vmd.gov.uk.
Queries related to all other (non-urgent) signal notification submissions should be directed either via VMDS to the PSUR Queries group or via e-mail to psur.queries@vmd.gov.uk.
3.1.4 Content of signal notifications
For signal notification, only certain fields within the ‘Benefit-risk statement’ sheet and ‘Signals and regulatory actions’ sheet need to be completed. MAHs do not need to complete the other sheets as these are only for use when submitting the annual BRR.
The following information should be completed by MAHs in the first sheet of the BRR template (further details on completion of these fields can be found within section 4.2 of this guideline):
-
Brand name
-
Marketing Authorisation Holder
-
Marketing Authorisation Number/Product Group Code
-
Submission type – here Signal notification of Urgent signal notification should be selected from the dropdown menu as applicable
No fields on the ‘Benefit-risk statement’ sheet of the template below the Submission type should be completed for a signal notification, these fields should only be completed for annual BRRs (see section 4 of this guideline).
The following information should be completed by MAHs in the ‘Signals and regulatory actions’ sheet of the BRR template:
-
signal VeDDRA preferred Term (or non-VeDDRA term if no suitable preferred Term)
-
species as per tab F. Species of the VICH GL30 Vocabulary Lists
-
date first detected
-
current status – ‘Ongoing’ should be selected from the dropdown menu unless the signal has already been closed by another regulatory authority
-
date closed (for closed signals)
-
source of signal (such as MAH database, regulatory authority database, literature report)
-
country (in which the signal was detected), using appropriate ISO-Country code as per ISO - ISO 3166 — Country Codes
-
evaluation and brief summary of the findings; information that should be considered for inclusion within this section may include:
*signalment and clinical details
-
brief description of event outcome
-
observed patterns of event development
-
details of other products
-
reversibility
-
supporting lab data
-
assessment of the causal relationship
-
details of regulatory procedures ongoing at the time of submission e.g. variations or signal processes, including those involving other regulatory authorities
-
details of the source
-
event incidence
-
for urgent signals in particular, an evaluation of the urgency and potential impact
-
proposed action
-
proposed action details
-
For the proposed action column, one of the following actions will need to be selected:
-
close monitoring
-
post-marketing surveillance study
-
variation, including change to the product literature
-
product/batch recall
-
suspension
-
withdrawal
-
risk management plan
-
no further action required
-
other
Only select ‘Other’ if no other option applies.
If close monitoring is proposed, the period over which the product will be closely monitored should be specified, alongside any details of additional monitoring processes that will be put in place.
If a post-marketing surveillance study is proposed then details regarding the scope, objectives, and timelines should be provided.
If a variation is proposed, details of this should be provided. If the variation proposed involves a change to the product literature, then details of the section of the product literature affected and proposed wording should be provided.
If a product/batch recall is proposed, details of the batches affected and whether they have been distributed should be provided.
If a suspension or withdrawal is proposed, details should be provided including dates if applicable.
If a risk management plan is proposed, details should be provided e.g. details of communications disseminated to specified stakeholder groups and a proposed timescale, or details of a restriction.
If the MAH proposes that no regulatory action should be taken, an explanation of why this has been proposed should be provided.
It is expected that for the majority of signals reported via a Signal notification during the reporting period, an action will be proposed, as a potential change to the benefit-risk balance will have been identified.
The VMD will contact the MAH for further information if required.
Final actions should be completed for ‘Closed signals’ only (where ‘Closed’ was selected for ‘Current status’). The majority of signals reported during the reporting period (as opposed to in the annual BRR) will be ongoing at the time of reporting and may require discussion with the VMD.
Signal management process overview
4. Benefit-risk reports
A BRR is an important document provided by the MAH to the VMD post-authorisation on an annual basis. The document is intended to provide an update on the worldwide benefit-risk balance of a product, including any validated signals detected that have previously been submitted to the VMD.
BRRs are required for all products authorised in the UK regardless of whether or not a product has been marketed. Electronic copies of the BRR template should be submitted in Excel format to the VMD using the Veterinary Medicines Digital Service(VMDS), a secure messaging service. Any MAH not signed up to VMDS can register at Veterinary Medicines Digital Service.
4.1 Benefit-risk report submissions
Following the placing of a VMP on the market in the UK, an MAH must submit a BRR once in the course of every year during the period of validity of the authorisation.
BRRs should be submitted regardless of whether the product was marketed or whether there were any signals detected throughout the year. They should cover the period of time since the data lock point (DLP) in the preceding BRR (or for first submission of a BRR, the final PSUR) and there should be no gaps in, or overlapping, of data.
The DLP should be no earlier than 2 months prior to the submission date.
It is recommended that the DLP date be on the last day of a month.
Any validated signals noted after the DLP but prior to submission of the BRR should be included in the subsequent BRR, unless they meet the criteria for an urgent or standard safety signal.
Submission dates for BRRs should align with the grouped active substance recommended due dates of submitting annual statements available on the EMA website.
For new Anatomical Therapeutic Chemical Classification System for veterinary medicinal products (ATCvet) codes which are not included in the list, or if there are any queries regarding ATCvet codes, MAHs should contact psur.queries@vmd.gov.uk.
The BRR should be submitted to the VMD no later than the applicable submission due date for the product.
In exceptional circumstances, MAHs can request to change the submission dates (and DLP) for the annual BRR by contacting psur.queries@vmd.gov.uk with a proposal for an alternative date, and an explanation of the reason for the request. Where possible, alternative dates should be the last day of a month.
BRRs should also be submitted immediately on request by the VMD on an ad hoc basis. The DLP and submission date for these ad hoc requests will be proposed by the VMD depending on the urgency of the issue and should be agreed with the MAH.
BRRs should be submitted as one completed Excel document per MA number or Product Group Code (see 4.3.3 of this guideline for further details) as per the BRR template.
The Excel document should be named using the applicable submission category date, MA number (with an underscore rather than forward slash separation) or Product Group Code (PGC) with an underscore rather than a dash, and document number 1 separated by underscores: YYYYMM_MAnumber_1 or YYYYMM_PGC_1. For a product with a submission category date of 31-AUG and an MA number 09285/8019 being submitted in the year 2024, for example, the name of the document would be 202408_09285_8019_1. For a product with a submission category date of 31 AUG and Product Group Code of GUOFGAND-BENEFITAS being submitted in the year 2024, for example, the name of the document would be 202408_GUOFGAND_BENEFITAS_8019_1.
Multiple BRR template signal documents can be submitted per VMDS submission as long as only one template is submitted per MA or PGC. If further information is requested or the MAH wishes to submit further documentation, then this can be added to the submission as a separate document.
Additional documents should be named using the applicable submission category date, MA number or PGC and subsequent document number (this would be 2 if two separate documents are submitted, and 3 if three separate documents are submitted and so on). For the 2nd document submitted for a product with a submission category of 31-AUG and an MA number 09285/8019 being submitted in the year 2024, for example, the title of the document would be 202408_09285_8019_2. The corresponding BRR document would have been named 202408_09285_8019_2.
Note that validation rules apply, so documents named using spaces or forward slashes rather than underscores, or other characters will be rejected automatically.
BRRs should be submitted using the Veterinary Medicines Digital Service (VMDS), a secure messaging service. Any MAH not signed up to VMDS can register at Veterinary Medicines Digital Service.
Once signed in to the VMDS account, the MAH should select the relevant group: PSUR Submission.
Queries related to BRR submission should be directed either via VMDS to the PSUR Queries group or via e-mail to psur.queries@vmd.gov.uk.
4.2. Content of benefit-risk reports
All BRRs should be completed in English; and all declarations and signals must take into account all adverse events arising in the UK and outside of the UK regardless of the route of authorisation.
It is strongly recommended that, before submitting the BRR, the MAH should make sure that all adverse event reports have been submitted electronically where required and duplicate detection performed.
The Benefit-risk report should either be submitted including sales data and worldwide authorisation status information as one complete document annually or can be split into multiple submissions.
If multiple submissions are submitted at different timepoints, one must contain at a minimum the benefit-risk statement and signals (if any have been detected during the reporting period) and be reported within 2 months of, and no later than, the appropriate submission date (unless otherwise agreed with the VMD in advance) and the remaining submission(s) must contain the sales data and worldwide authorisation status between them, and be received at least once annually. Multiple sales data submissions can be made throughout the year, as long as there are no gaps in data and total full data has been provided at least once per annum.
If Sales data or Worldwide authorisation status data are submitted outside of the BRR submission, they must be submitted using the entire BRR template (including all sheets, even if some sheets are unused). The Brand name, Marketing Authorisation Number/Product Group Code and Submission type on the Benefit-risk statement should be the only other fields completed in addition to one or more of the Worldwide authorisation status, UK sales data and Third country sales data sheets.
For pharmacovigilance data to be correctly attributed to the right product in the VMD databases MAHs must use an up to date MA number or PGC. If a product authorised in the UK has multiple MA numbers due to a splitting of a previous MA number into GB and NI MA numbers, either 2 separate submissions should be made, one for each MA number or a PGC will need to be created.
The following sections will provide details on how to fill out the BRR template.
4.3 Benefit-risk statement
The first sheet in the BRR template is the benefit-risk statement.
For an annual BRR submission MAHs should complete:
-
Brand name
-
Marketing Authorisation Holder
-
Marketing Authorisation Number/Product Group Code
-
Submission type – here Annual benefit-risk report should be selected from the dropdown menu
-
Data Lock Point
-
Period of report from
-
Period of report to
-
Have there been any validated signals during the reporting period?
-
Benefit-risk statement
4.3.1 Brand name
For BRRs involving a single MA number, this should be identical to the name of the VMP as stated in the national MA and associated documentation.
For BRRs involving multiple MA numbers, the group name forming the second part of the Product Group Code should be used (see section 4.3.3 of this guideline for further details on Product Group Codes).
4.3.2 Marketing Authorisation Holder
This should be identical to the business name of the MAH as stated in the national marketing authorisation and associated documentation.
4.3.3 Marketing Authorisation Number/Product Group Code
For BRRs involving a single MA, this should be identical to the MA number as stated in the national MA and associated documentation, unless the MA number has been updated since initial authorisation, in which case the current MA number must be used.
If a national authorisation number is used, enter only the authorisation number itself within the Excel spreadsheet, without Vm or Vh preceding it.
For BRRs involving multiple MAs, a Product Group Code (PGC) must be used. The PGC links a set of MAs together into a single group and should be used to link both UK and third country products.
This must be either proposed by the MAH and accepted by the VMD or requested by the MAH at least one month prior to the first BRR submission, and a BRR should not be submitted until this Product Group Code has been agreed.
A MAH must either:
-
Propose:
a) A list of MA numbers and their associated brand name and authorisation country grouped by PGC
b) One or more PGCs following the below naming convention
c) The ATCvet code submission due date by which this PGC will be submitted
-
Provide (in order for the VMD to create a PGC):
a) A list of MA numbers and their associated brand name and authorisation country grouped by PGC
b) The ATCvet code submission due date by which this PGC will be submitted
-
Provide (if no PGC is required):
a) A list of MA numbers which will be submitted individually and their brand names
b) The ATCvet code submission due date by which each MA will be submitted
The group must contain only similar VMPs which originate from the same MAH being responsible for pharmacovigilance of these VMPs, have the same active ingredients (substances), have the same major excipients with the same or similar pharmaceutical function and at least one common registered species.
In some cases it will only include different strengths of the same product. Product groupings that are considered too large or diverse may be declined by the VMD and an alternative suggestion made.
Note that MAHs with products that have split GB/NI MA numbers will need to request a Product Group Code as only one MA number can be submitted per BRR.
The same product group should be intended to be used for all future submissions. The VMD must be contacted if the MAH wishes to add products (for example, a newly authorised product), remove products (for example, a product no longer authorised or moved under the responsibility of a different MAH) or alter products within this grouping.
The Product Group Code should take the following format:
MAHORGID-GROUPNAME
The MAHORGID is the 8-digit organisational ID (used as part of your worldwide case number (AERID)) used for adverse event case submission. The group name must be an overarching product name for all the MAs within that PGC.
If the MAGHORID does not reflect the MAH name clearly an alternative to the MAGHORID can be proposed, but this ID should be no longer than 8 characters in length.
For example, if the VMD were to have a Product Group Code for the invented products Benefitas 10 mg and Benefitas 20 mg, with a chosen overarching product name of Benefitas, the PGC would be VMDDEFRA-BENEFITAS.
Initial Product Group Code/MA/ATCvet code information should be sent to psur.queries@vmd.gov.uk with the subject ‘BRR group’.
Ongoing queries related to Product Group Codes should be directed either via VMDS to the PSUR Queries group or via e-mail to psur.queries@vmd.gov.uk.
4.3.4 Annual benefit-risk report or signal notification
Select ‘Annual benefit-risk report’.
4.3.5 Data lock point
The DLP should be entered DD/MM/YYYY, and must be within the 2 months prior to the submission due date.
4.3.6 Period of report from
The start date of the reporting period covered by the annual BRR in the format DD/MM/YYYY.
4.3.7 Period of report to
The end date of the reporting period covered by the annual BRR in the format DD/MM/YYYY.
4.3.8 Validated signals
Select ‘Yes’ or ‘No’ from the dropdown menu as applicable for ‘Have there been any validated signals during the reporting period?’
Enter Yes if any signals have been included in the Signals and regulatory actions sheet, or have been already submitted during the reporting period.
4.3.9 Benefit-risk statement
The MAH should select ‘Yes’ or ‘No’ from the dropdown menu as applicable in response to the statement, ‘The benefit-risk balance of the product remains the same and no action is required.’
If a MAH selects ‘No’ as a signal has been noted that alters the benefit-risk balance of the product or further information is required to determine this via, for example, close monitoring or a post-marketing surveillance study, then this should be detailed in the ‘Signals and regulatory actions’ sheet of the BRR.
4.4 Backlog line listings
Any non-serious UK cases not previously submitted to the VMD i.e. since last PSUR DLP, can either be submitted electronically or should be included in a line listing in the relevant sheet ‘Backlog line listings’ of the BRR/Signal template.
All third country cases not previously submitted to the VMD since the last PSUR DLP should also be added to this tab, if not submitted electronically.
In subsequent BRRs this sheet does not need to be completed as all adverse event cases should be sent electronically within 30 calendar days of receipt by the MAH to the VMD.
Guidance on the required information for adverse event reports, and particular types of reports can be found in section 4 of Guideline III Adverse event reporting.
The following fields must be completed if the information is available.
Country (2 character country codes ISO 3166)
The country where the adverse event occurred should be entered in 2 character country code format using the appropriate ISO-Country code as per ISO - ISO 3166 — Country Codes.
Unique Adverse Event Report Identification Number (Worldwide Case Number)
The Unique Adverse Event Report Identification Number (sometimes known as AERID or the worldwide case number) is electronically assigned by the first organisation to send an adverse event report.
This number consists of:
-
country of occurrence code (3 characters using appropriate ISO-Country code as per tab A.36 ISO-Country of the VICH GL30 Vocabulary Lists)
-
the MAH’s organisation ID (MAHORGID,8 characters)
-
remaining free text (up to 47 characters, which can include a routing ID)
Original Receive Date
The date of first communication of an adverse event report from the primary reporter to the MAH or regulatory authority. It should be entered in the format DD/MM/YYYY.
Date of First Exposure
The date of first exposure/treatment with the product involved in the adverse event. It should be entered in the format DD/MM/YYYY.
Date of Onset of Adverse Event
The date of onset of the adverse event. It should be entered in the format DD/MM/YYYY.
Number of Animals Treated
The number of animals treated or humans exposed.
For human exposure cases, this will always be 1 as there should only ever be one human per adverse event report.
Species
Species should be entered as per VICH GL30. Enter ‘Human’ for human adverse event reports.
Breed
Breed should be entered if known, using the most relevant available breed from VICH GL30.
Enter N/A for human adverse event reports.
Age
This should be entered in years.
Number of Animals Affected
The number of animals/humans affected in the adverse event report, including indirectly exposed animals, for example, those treated during pregnancy or lactation, co-mingled, or via infectious spread.
This should always be entered as 1 for human adverse event reports.
Number of Animals Died
The number of animals/humans for which the outcome of the adverse event was death.
Use According to Label
For animal cases, this should be entered as ‘Yes’ for use in accordance with the MA, or ‘No’ for use outside the terms of the MA.
For human adverse event reports, N/A should be entered unless the case involves deliberate misuse by a human which has led to adverse events in a human, in which case enter ‘No’.
Other products administered
List all other products that have been added to the products section of the case, separated by commas. Brand names or active ingredients of products can both be added.
VeDDRA Low Level Terms
Add all VeDDRA LLTs that have been added to the case, separated by commas.
Narrative of Adverse Event
A description of the adverse event should be provided. The recommended content of adverse event narratives is covered in Guideline III Adverse event reporting.
4.5 Worldwide authorisation status
Worldwide authorisation status data can either be submitted as part of the annual Benefit-risk submission or at a different timepoint, as long as at least one submission is received annually.
If data are submitted outside of the annual BRR submission, they must be submitted using the entire BRR/Signal template. The Brand Name, Marketing Authorisation Number/Product Group Code and Submission type on the Benefit-risk statement should be the only other fields completed in addition to the Worldwide authorisation status sheet.
If Worldwide authorisation status and Sales data are submitted at the same timepoint but separate from the annual BRR submission, select Sales and worldwide authorisation status from the dropdown list for Submission type.
Further details on submission of worldwide authorisation status data can be found in section 4.6 of this guideline.
Provide the authorisation status per country, including the authorisation number and whether the product is marketed in that country or not (since the time of the last BRR submission). Complete ‘If marketed in the UK since last BRR: date first marketed’ and ‘If withdrawn from UK market since last BRR: date withdrawn from the market’ if applicable with dates in the format in the format DD/MM/YYYY.
Providing marketing status for every country is not mandatory, however should be included for the countries for which it is available.
This should be completed for all countries, including the GB/NI.
The MAH should complete the following fields:
Country (2 character country codes ISO 3166)
The country where the product is authorised should be entered in 2 character country code format using the appropriate ISO-Country code as per ISO - ISO 3166 — Country Codes.
Brand Name
This should be identical to the name of the VMP as stated in the MA for the applicable country.
Marketing Authorisation Number
This should be identical to the MA number as stated in the MA for the applicable country. If there is no MA number or other identifier, this can be left blank.
Marketed
Select ‘Yes’ or ‘No’ from the dropdown menu depending on whether the product is currently marketed in the applicable country. If this information is not available for one or more countries, UNK should be selected for these countries.
If marketed in the UK since last BRR: date first marketed
Only complete if the product has first been marketed in the UK since the last annual BRR submission. Enter the date in the format DD/MM/YYYY. Leave blank if this does not apply.
If withdrawn from UK market since last BRR: date withdrawn from the market
Only complete if the product has first been marketed in the UK since the last annual BRR submission. Enter the date in the format DD/MM/YYYY. Leave blank if this does not apply.
4.6 Sales data and submission of standalone sales/worldwide authorisation status data
Sales submissions (either as part of the annual Benefit-risk report or submitted at a different timepoint) should contain one or more of the following:
- Sales data for the UK as a whole (not separated into GB/NI)
- Sales data for EEA countries (listed individually)
- Combined sales data for non-EEA countries (total for all non-EEA countries grouped together)
Multiple sales data submissions can be made throughout the year, as long as there are no gaps in data and total full data for UK, EEA and non-EEA has been provided at least once per annum.
Sales data for the UK should be provided for all each pack sizes covered by each MA.
Sales data for EEA and non-EEA countries should be provided for all pack sizes covered by each MA, or per Product Group Code (for further details see section 4.6.4).
Queries related to selection of the appropriate sales volume unit, pack unit size or dose factor should be directed to psur.queries@vmd.gov.uk.
If sales data are submitted at any time other that as part of the annual Benefit-risk report submission, they should be submitted to the VMD via the Veterinary Medicines Digital Service (VMDS). Any MAH not signed up to VMDS register at Veterinary Medicines Digital Service - GOV.UK (www.gov.uk).
Once signed in to the VMDS account, the MAH should select the relevant group: Sales and Worldwide Authorisation Status.
The entire Excel document should be submitted with the relevant sections completed as detailed in sections 4.2, 4.6.3 and 4.6.4 of this guideline. It should be named using the applicable MA number (with an underscore rather than forward slash separation) or Product Group Code (PGC) with an underscore rather than dash separation, the year of submission, type of submission (Sales, WWA or SalesWWA) and number describing the order of sales submitted, separated by underscores:
MAnumber_YYYY_Sales_x or MAnumber_YYYY_WWA_x or MAnumber_YYYY_SalesWWA_x
PGC_YYYY_Sales_x, or PGC_YYYY_WWA_x or PGC_SalesWWA_x
Sales should be used for standalone sales data only, WWA for standalone worldwide authorisation status data only, and SalesWWA if both sales and worldwide authorisation status data is submitted at the same time.
Number 1 should be used for the first sales +/- WWA submission for that product/PGC submitted in that year, and 2, 3, 4 etc used for subsequent submissions. For example, the first standard standalone sales document for a product with an MA number 09285/8019 being submitted in the year 2024, would be named 09285_8019_2024_Sales_1. The second would be 09285_8019_2024_Sales_2.
It is not anticipated that additional documents other than the Excel document will be submitted. If further documentation is considered necessary, contact the VMD prior to sending.
Queries related to all other sales +/- worldwide authorisation status submissions should be directed either via VMDS to the PSUR Queries group or via e-mail to psur.queries@vmd.gov.uk.
4.6.1 Sales volume units
The following should be used for sales volume units:
-
vaccines to be expressed in doses
-
liquid to be expressed in litres
-
powder to be expressed in kilograms
-
tablets to be expressed in numbers of tablets
-
sprays to be expressed in litres or kilograms
-
collars to be expressed in numbers of collars
-
paste to be expressed in kilograms
-
pipettes for spot-on solution to be expressed in vials
-
Additional units that may be used include boluses, capsules, implants, patches, sponges, and strips.
If a single MA covers multiple presentations of a product e.g. 5ml and 10 ml syringe sizes, the sales volume should be expressed in litres. Selecting a unit e.g. syringes would not be appropriate.
If a single MA covers multiple presentations of a product that cannot be effectively expressed in litres/kilograms e.g. 2 separate collar sizes, details should be provided (see section 4.6.2 of this guideline).
4.6.2 Dose factor
The dose factor is a positive numerical value which provides an estimate of the number of treated animals using the sales data. It should equal the average number of animals of a target population which can be treated by one package of a given pack size of a product, regardless of the formulation. It should be calculated independently of reported adverse events. A separate dose factor should be calculated and submitted for each marketed pack size. Dose factors for pack sizes that are not marketed at the time of BRR submission do not need to be included.
If multiple pack sizes contain the same amount of individual units e.g. a tub of 250 tablets and a pack of 50 blister strips containing 5 tablets per strip, only one pack size (and therefore dose factor) need be submitted.
As much as possible, it should be kept consistent across reporting periods to allow comparability of incidence calculations, and ideally be kept consistent between reference and generic products for which the MAH has responsibility. Any changes to the dose factor should be highlighted and justified within the related BRR.
The calculation of an appropriate dose factor will depend on factors such as the type of product, target species, production status, formulation, indication and treatment regimen, and should be determined by the MAH. Any suggestions for methods for calculation should only be used if deemed representative of the conditions of use of the product.
For VMPs that are administered as a single dose, where the pack unit amount is 1 dose, the number of doses equals the number of animals treated and the dose factor would be 1.
Examples include flea collars, and some vaccines, long-acting injectable preparations, wormers and flea spot-ons.
For VMPs where one unit of product is administered for the entire course of treatment for an individual animal, the number of units again equals the number of animals treated (1 unit = 1 animal treated), and therefore the dose factor would be 1.
Examples include VMPs for topical use, such as shampoos, pastes, eye preparations or ear preparations. For other single dose administration VMPs, a general suggested formula is to divide the package volume by the single dose amount (e.g. 1 dose or 10ml).
For single dose administration VMPs where there is a range of doses that could be administered as per the SPC, the dose factor should be calculated using the maximum dose in this range; divide the package volume by the single maximum dose amount (e.g. 1 dose or 10ml).
If an active substance concentration is applicable to the product:
- Multiply the concentration of the active substance (in mg/ml or mg/mg) as stated in the product literature by the package volume (ml, g)
- Multiply the weight of the target species (derived from the table provided below) by the maximum dose (in mg/kg or ml/kg) as stated in the product literature
- Divide the result of step 1 by step 2
For non-single dose administration VMPs where there is a set dose and duration of treatment that should be administered as per the SPC:
- Multiply the recommended dose based on standard weight (in mg, ml, tablet etc) as stated in the product literature by the duration of treatment (days)
- Divide the package volume by the result of step 1.
For non-single dose administration VMPs where there is a range of doses and durations that could be administered as per the SPC, the dose factor should be calculated using the maximum dose in these ranges, for example:
- Multiply the recommended maximum dose based on standard weight (in mg, ml, tablet etc) as stated in the product literature by the maximum duration of treatment (days)
- Divide the package volume by the result of step 1
For non-single dose administration VMPs where there is a range of doses and durations that could be administered as per the SPC and If an active substance concentration is applicable to the product:
- Multiply the package volume by the concentration of the active substance (in mg/ml or mg/mg) as stated in the product literature.
- Multiply the weight of the target species (derived from the table provided below) by the maximum dose (in mg/kg or ml/kg) and by the maximum duration (days) as stated in the product literature
- Divide the result of step 1 by the result of step 2
For VMPs which are indicated for continuous long-term/lifelong treatment, the maximum estimated dose per animal over a 6-month period should be used (utilising a maintenance dose stated in the SPC if applicable) i.e. maximum estimated dose (mg, ml, g, tablet etc) x 182 days.
For VMPs which are indicated for both short-term and long-term treatment, where there is no defined length of treatment, the maximum estimated dose per animal over a 1-month period should be used i.e. maximum estimated dose (mg, ml, g etc) x 30 days.
In general, where there is a range provided in the Summary of Product Characteristics (SPC) for the dose and/or duration, use the maximum recommended dose and/or longest duration of treatment as applicable. Any other dose/duration should be justified.
For VMPs which are used for multiple animals, and contain multiple doses per package:
- Multiply volume of a single dose (e.g. litres) by the duration (days) of the treatment
- Divide the result of step 1 by the estimated dose (e.g. litres)
- Divide the total pack size (total volume) by the result of step 2.
For dry cow intramammary products, 1 dose is considered equivalent to 4 intramammary syringes, and for lactating cow intramammary products, 1 dose is considered equivalent to 1 intramammary syringe.
For inhalation anaesthetics, a duration of anaesthesia of 45 minutes at the typical rate used for maintenance should be used.
The average weight of the target species or population should be derived from the table below.
If there is a range of weights or production status within a target population e.g. use in newborns as well as adults, an average weight should be determined based on the estimated use within a species group.
If there is a range of species within a target population and the species are defined, separate dose factors per species can be listed within the Additional Information cell.
If there is a range of species within a target population where the species split cannot be defined (e.g. ‘poultry’), an average weight should be determined based on the estimated use within the target population, calculated by estimated species split alongside estimated production status within the individual species.
A brief explanation should be provided, including the estimated percentage of each species use.
If no dose factor is provided by an MAH, the VMD will determine a dose factor based on the product, previous PSURs/Benefit-risk reports and other relevant data.
| Species, sub-population | Standard weight (Kg) |
|---|---|
| Dog | 20 |
| Cat | 5 |
| Rabbit | 1.5 |
| Guinea pig | 1 |
| Ferret | 1.4 |
| Horse | 550 |
| Adult cow | 550 |
| Beef calf | 150 |
| Newborn calf | 50 |
| Sow/boar | 160 |
| Breeding sow | 240 |
| Finishing pig | 60 |
| Weaner pig | 25 |
| Piglet | 2 |
| Adult sheep/goat | 60 |
| Breeding sheep/goat | 60 |
| Sheep/goat under 12 months | 20 |
| Lamb/kid | 10 |
| Chicken, broiler | 1 |
| Chicken, layer | 2 |
| Turkey | 10 |
| Turkey, poult | 4 |
| Duck | 2 |
| Goose | 5 |
| Pheasant, Guinea fowl | 0.5 |
| Salmon | 3 |
Exposure in pigeons is recommended to be calculated on the basis of 30 pigeons/litre of drinking water. The dose factor should be submitted as a numerical value with up to 4 decimal digits only.
A brief explanation of the justification for the dose factor should be provided within the ‘Explanation’ field. If more extensive information needs to be provided, such as calculations, this can be submitted alongside the BRR as a separate document. If a separate document is submitted, this should be specified by selecting ‘Yes’ on the Benefit-risk statement page.
Incidence % should be calculated for signals submitted, using the following formula:
Incidence % = (Number of animals reacted ÷ number of animals treated) x 100
The number of animals treated should be calculated using the dose factor (sales volume x dose factor). The number of animals reacted should include all on and off-label events including lack of efficacy and N-causality events.
4.6.3 UK sales data
The MAH should complete the following fields per pack size per MA number:
Marketing Authorisation Number
This should be identical to the MA number as stated in the national MA and associated documentation, unless the MA number has been updated since initial authorisation, in which case the current MA number must be used.
If a national authorisation number is used, enter only the authorisation number itself within the Excel spreadsheet, without Vm or Vh preceding it.
If a MA number has been split into GB and NI numbers, list only the GB number in this field, and add the NI number to the Additional information field.
Country (2 character country codes ISO 3166)
The country where the product is marketed should be entered in 2 character country code format using the appropriate ISO-Country code as per ISO - ISO 3166 — Country Codes.
Month, Year
The breakdown of sales data submitted should be per month. If monthly figures for sales data are unknown, an estimate should be calculated from the shortest time period available to provide the most accurate data. For example, if both 3 monthly and 12 monthly sales data breakdowns are available, the 3 monthly data should be divided by 3 to achieve the estimated monthly sales, rather than using the 12 monthly sales data.
Year should be entered in the format YYYY.
Month should be entered in the format MM.
Full months of data should be submitted. There must be no gap in reporting of sales data.
Period 2 would cover the remaining sales data from the previous calendar year and period 1 would cover the sales data from the current calendar year, up to until the reporting period end date.
Pack unit size
The pack unit size is the total number of units within a package size e.g for a tub of 100 tablets, this would be a numerical value of 100, and for a blister pack of 50 strips with 5 tablets per strip, this would be 250.
Number of pack units
Enter the numerical value of the number of pack units. The pack unit should be equivalent to that used in the dose factor calculation. If no pack units were sold during the period (for a country in which the product is authorised), enter 0. If further explanation of the pack unit is necessary, enter further details within the Additional information field.
Dose factor
This should be entered as a numerical value, with no more than 4 decimal digits. See section 4.6.2 of this guideline for further information on dose factor calculation.
Sales volume
Enter the numerical value of the volume of sales. If no sales occurred during the period (for a country in which the product is authorised), enter 0.
Units
Select the appropriate unit from the dropdown menu, aligning with section 4.6.1 of this guideline.
Estimated monthly
If the monthly breakdown of sales data is unknown, and therefore an estimate has been calculated, select Yes. If monthly data is known, select No.
Estimated GB/NI split
For sales data where an accurate split between GB and NI data is known and can be provided, enter No and provide details within the Additional Information field. Provision of this data is not a mandatory requirement.
If the split between GB and NI data can be estimated, enter Yes and provide details within the Additional Information field. If this data is unknown, is not applicable (the product is marketed either solely in GB or NI) or not provided for any other reason, enter N/A and provide brief clarification within the Additional Information field.
Additional information
A brief explanation of the justification for the dose factor should be provided. If more extensive information needs to be provided, such as calculations, this can be submitted alongside the BRR as a separate document.
If a single MA covers multiple presentations of a product that cannot be effectively expressed in litres/kilograms e.g. 2 separate collar sizes, the number of each unit type making up the sales volume should be stated, and whether this is an estimated figure or not. A brief justification for the dose factors used should additionally be entered here.
Details of NI MA numbers (when a GB/NI MA number split has occurred) and estimated GB/NI sales split details should be entered in this field as explained above under the heading ‘Estimated’.
4.6.4 Third country sales data
Third country sales data can be provided in one of the following formats:
- Pack unit size, Number of pack units, Dose factor, Sales volume, Units per MA number
- Sales volume and units only per Product Group Code
- Sales volume and units only per MA
The MAH should complete the following fields per pack size per MA number for each third country within the EEA, and as grouped data for non-EEA third countries:
Marketing Authorisation Number
This column should contain either the MA number as stated in the MA for the applicable country or Product Group Code. If there is no MA number or other identifier, and a Product Group Code is no applicable, this can be left blank.
Country (2 character country codes ISO 3166)
The country where the product is marketed should be entered in 2 character country code format using the appropriate ISO-Country code as per ISO - ISO 3166 — Country Codes. For non-EEA country grouped data, Non-EEA should be entered.
Month, Year
The breakdown of sales data submitted should be per month. If monthly figures for sales data are unknown, an estimate should be calculated from the shortest time period available to provide the most accurate data. For example, if both 3 monthly and 12 monthly sales data breakdowns are available, the 3 monthly data should be divided by 3 to achieve the estimated monthly sales, rather than using the 12 monthly sales data.
Year should be entered in the format YYYY.
Month should be entered in the format MM.
Full months of data should be submitted. There must be no gap in reporting of sales data.
Number of pack units
Enter the numerical value of the number of pack units. The pack unit should be equivalent to that used in the dose factor calculation. If no pack units were sold during the period (for a country in which the product is authorised), enter 0. If further explanation of the pack unit is necessary, enter further details within the Additional information field.
This may be left blank if only sales volume and unit data is submitted.
Pack unit size
The pack unit size is the total number of units within a package size e.g for a tub of 100 tablets, this would be a numerical value of 100, and for a blister pack of 50 strips with 5 tablets per strip, this would be 250.
This may be left blank if only sales volume and unit data is submitted.
Dose factor
This should be entered as a numerical value, with no more than 4 decimal digits. See section 4.6.2 of this guideline for further information on dose factor calculation.
This may be left blank if only sales volume and unit data is submitted.
Sales Volume
Enter the numerical value of the volume of sales. If no sales occurred during the period (for a country in which the product is authorised), enter 0.
Units
Select the appropriate unit from the dropdown menu, aligning with section 4.6.1 of this guideline.
Estimated monthly
If the monthly breakdown of sales data is unknown, and therefore an estimate has been calculated, select Yes. If monthly data is known, select No.
Estimated GB/NI split
Enter N/A for third country sales.
Additional information
A brief explanation of the justification for the dose factor should be provided. If more extensive information needs to be provided, such as calculations, this can be submitted alongside the BRR as a separate document.
If a single MA covers multiple presentations of a product that cannot be effectively expressed in litres/kilograms e.g. 2 separate collar sizes, the number of each unit type making up the sales volume should be stated, and whether this is an estimated figure or not. A brief justification for the dose factors used should additionally be entered here.
4.7 Signals and regulatory actions
All validated signals which following assessment are deemed to not suggest a new risk or change to the benefit-risk balance (all validated signals that have not previously been submitted to the VMD via a Signal notification), should be submitted annually via the BRR/Signal template. Applicable signals that have been assessed via other regulatory authorities should also be included within this section.
Non-validated signals should be recorded by the MAH but should not be submitted to the VMD.
Signals should be listed within the ’Signals and regulatory actions’ sheet of the BRR.
Note that the VMD should be additionally notified of validated signals where there is a potential risk or change to the benefit-risk balance within 30 calendar days, or no later than the next working day for urgent safety signals the require restrictions to be implemented or 3 days where no restrictions are deemed necessary.
The following information should be completed by MAHs in the ’Signals and regulatory actions’ sheet of the BRR/Signal template:
VeDDRA preferred term (or non-VeDDRA term if there is no suitable preferred term)
A separate row should be used for each individual VeDDRA PT or non-VeDDRA term.
Species
Species should be entered as per VICH GL30. Enter ‘Human’ for human adverse event reports.
The same signal affecting a different species should be entered on a separate row (one row per species affected).
Date signal detected
The date the signal was first detected by the MAH’s internal signal management processes or by a regulatory authority, whichever occurred first. Enter in the date format DD/MM/YYYY
Current status
Select ‘Ongoing’ or ‘Closed’ from the dropdown menu.
‘Closed’ should only be selected for any signal where the action has been completed, or for signals where it is proposed or has been agreed by a regulatory authority that there should be no further action.
Date closed
The date that the action taken was finalised. This would be the date that a variation was completed, or a product/batch was recalled/suspended/withdrawn. This should be entered in the format DD/MM/YYYY.
Source of signal
Suggestions for sources include MAH database, regulatory authority database, literature report.
Country
The country where the signal was noted should be entered using appropriate ISO-Country code as per ISO - ISO 3166 — Country Codes. If the signal is not country-specific, this field can be left blank but any applicable location-related information should be provided within the ‘Evaluation and summary of findings’ field.
Evaluation and brief summary of the findings
This should provide adequate evidence of how the signal was validated and assessed. Information that should be considered for inclusion within this section may include:
-
Signalment and clinical details
-
brief description of event outcome
-
location related data if non-country specific
-
observed patterns of event development
-
details of other products
-
reversibility
-
supporting lab data
-
assessment of the causal relationship
-
details of regulatory procedures ongoing at the time of submission e.g. variations or signal processes, including those involving other regulatory authorities
-
details of the source
-
event incidence
-
an evaluation of the potential impact
Proposed action
This is the action that was initially proposed by the MAH or regulatory authority.
Select the appropriate action from the dropdown menu. The options available are:
-
close monitoring
-
post-marketing surveillance study
-
variation, including change to the product literature
-
product/batch recall
-
suspension
-
withdrawal
-
risk management plan
-
no further action required
-
other
Only select ‘Other’ if no other option applies.
Proposed action details
Provide further details of the proposed action.
If close monitoring is proposed, the period over which the product will be closely monitored should be specified, alongside any details of additional monitoring processes that will be put in place.
If a post-marketing surveillance study is proposed then details regarding the scope, objectives and timelines should be provided.
If a variation is proposed, details of this should be provided. If the variation proposed involves a change to the product literature then details of the section of the product literature affected and proposed wording should be provided.
If a product/batch recall is proposed, details of the batches affected and whether they have been distributed should be provided.
If a suspension or withdrawal is proposed, details should be provided including dates if applicable.
If a risk management plan is proposed, details should be provided e.g. details of communications disseminated to specified stakeholder groups and a proposed timescale, or details of a restriction.
If the MAH proposes that no regulatory action should be taken, an explanation of why this has been proposed should be provided.
Final action
This should be completed for closed signals only (where ’Closed’ was selected for ‘Current status’.
It should be left blank for ongoing actions, such as a post-surveillance study, risk management plan or close monitoring. If these actions were both carried out as proposed and a resulting final action was also completed within the reporting period (such as a variation or a new decision made that no further action is required as a result of the proposed action), this resulting final action should be entered.
Select from the dropdown menu one of the following:
-
variation completed
-
product/batch recall
-
suspension
-
withdrawal
-
no further action required
-
other
Only select ‘Other’ if no other option applies.
Final action details
Provide further details of the final action.
If a variation, such as a change to the product literature, was initially proposed, and a variation has since been completed, details of the section of the product literature affected and finalised wording should be provided.
If a product/batch recall has been carried out, details of the batches affected, whether they have been distributed should be provided.
If a suspension or withdrawal is proposed, details should be provided including dates if applicable.
If ‘No further action required’ is selected, an explanation should be provided, unless this matches the initial proposed action and therefore an explanation has already been provided within the proposed action details section.
The VMD will contact the MAH for further information if required.
4.8. BRR assessment
If the VMD has any comments or questions related to the BRR submission, the VMD will inform the MAH of these via a BRR assessment report. Responses to questions should be addressed by the MAH within 60 calendar days. Any changes or variations requested should be submitted within 6 months unless otherwise requested or agreed.
Changes may be requested for any section of the product literature.
Requests for changes to the product literature should be made in line with the appropriate GB QRD template.